
1. INTRODUCTION
Sex determination has received much research attention in various species. Mollusca is the second largest phylum of invertebrates, which exhibits different reproductive strategies, including gonochoric, hermaphroditic, and sexual reversal.1 As a class of mollusks, all species of cephalopods are strictly gonochorism,2 the molecular regulation mechanism of sex determination may be relatively simple. The common long-arm octopus, Octopus minor (Cephalopoda: Octopodidae) is widely distributed on the tidal flats along the coast of China, Korea and Japan.3 As a kind of important shellfish resource in South Korea and the north of China,4 O. minor has become a commercially and ecologically important species, and one of the most widely studied octopods in east Asia.5 Due to Generational cycle and small body size, O. minor could be regarded as a model species to study the molecular regulation mechanism of sex formation in mollusks.
Sex formation is triggered by two major types of sex regulating factors (genetic and/or environmental factor) and the cascaded signal pathway of genes related to sex differentiation makes bisexual organisms that show sex dimorphism in phenotype nature.6 RNA-Seq was proved to be effective to reveal gene expression in specific tissues and organs, and gonad transcriptome sequencing has been commonly applied in screening gonadal differentiation genes.7 Some studies have investigated the gene regulations of sex determination in mollusks, and several genes have been identified to be sex-related, such as doublesex (DSX), SH3 domain-containing kinase-binding protein 1 (SH3KBP1), double-sex and mab-3-related transcription factor 1 (DMRT1), forkhead transcription factor gene 2 (Foxl2), gonadotropin-releasing hormone (GnRH), proliferation cell nucleus antigen (PCNA), β-catenin, transcription factor SoxE (SoxE) and transcription factor SoxH (SoxH),1,8–11 while similar studies are rare in octopus, especially in O. minor.
In the present study, we investigated the transcriptional profiles of male and female individuals at two gonad development stages in common long-arm octopus O. minor. Candidate genes related to sex regulating in O. minor were screened and discussed. The purpose of this article was to identify potentially sex regulating genes in O. minor which would provide information for further research on the regulatory mechanisms of sex determinisms and provide candidate genes for developing biotechnology in large-scale production of all-male O. minor.
2. MATERIALS AND METHODS
All experimental treatments involved in animal collection, rearing and dissection were implemented according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The study protocol was approved by the Experimental Animal Ethics Committee, Yantai Marine Economic Research Institute, China.
2.1. Animal sampling
Adult octopus (n=33) were collected in July and November from the coastal area of Muping, Shandong, China. The octopus were acclimatized in indoor concrete tank with sand-filtered seawater for three days. The octopus were anesthetized with 7.5% magnesium chloride according to Zhu et al.,12 and gonad tissues were then sampled individually. A portion of each gonad sample was frozen immediately in liquid nitrogen and stored at -80°C for following RNA extraction. The remaining part was stored in Bouin’s liquid for sexuality and gonad development stage determination by histologic section. Finally, 12 gonad samples of 4 groups i.e. mature male (MM), immature male (IMM), mature female (MF), immature female (IMF), were obtained according to their sexuality and gonad development stage.
2.2. RNA extraction, library construction and sequencing
The tissue was ground in a mortar under the condition of liquid nitrogen to get homogenate. Total RNA was extracted from each sample using TRIzol reagent (Invitrogen, CA, USA) following the manufacturer’s instructions and genomic DNA were removed using DNase (TaKaRa). The purity and concentration of RNA were determined using a Nano-Drop2000 spectrophotometer (Thermo, USA), and the integrity was determined by 1% agar-gel electrophoresis and Agilent 2100 (Agilent Technologies, Co. Ltd., USA). A total of 1 μg RNA per sample was used as input material for the library construction. The NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA) were used to generate the library according to the manual. And then, RNA was sequenced on Illumina HiSeq 2500 for 125 bp paired-end reads at Biomarker Biotechnology Corporation (Beijing, China).
2.3. Sequencing data processing
The raw data were firstly processed by removing reads containing adapter, ploy-N sequences and low quality reads through in-house Perl scripts. At the same time, Q20, Q30 and GC-content of the clean data were calculated for quality control. These high-quality clean data were aligned to the O. minor genome using Hisat2 (v2.0.4).13 Only reads with a perfect match or one mismatch were further analyzed and annotated based on the reference genome. Then, the mapped reads were assembled using Stringtie software (v2.1.4),14 and identified novel genes by comparing with the original genome annotation information. The novel genes were annotated using the public databases, including Nr (NCBI non-redundant protein sequences), Nt (NCBI non-redundant nucleotide sequences), Pfam (Protein family), KOG/COG (Clusters of Orthologous Groups of proteins), Swiss-Prot (A manually annotated and reviewed protein sequence database), KO (KEGG Ortholog database) and GO (Gene Ontology).
2.4. Identification and enrichment analysis of differentially expressed genes (DEG)
FPKM, fragments per kilobase of transcript sequence per million base pairs sequenced,15 were used to quantify gene expression level. The FPKM and differential expression analysis were performed using the R statistical software DESeq2 (1.18.0). The log2(fold change) > 1 and adjusted P-value < 0.01 were set as the threshold to judge the differentially expressed genes. Functional enrichment analysis of DEGs was then conducted. Gene Ontology (GO) enrichment analysis of the DEGs was implemented by the GOseq R packages based Wallenius non-central hyper-geometric distribution 16. KEGG enrichment analysis was implemented by KOBAS 17 software. P value <0.05 was used as the screening criterion for significant enrichment.
2.5. Validation through quantitative RT-PCR
Ten DEGs were randomly selected for qRT-PCR analysis to verify the RNA-Seq results. Primer Premier 5 software was used to design the primers for qRT-PCR (Table. S1). U6 was used as the internal reference gene. The cDNA synthesis was carried out with the reverse transcription Kit (TUREscript 1st Stand cDNA SYNTHESIS Kit) according to the manufacture’s introductions. qRT-PCR assay was performed using analytikjena-qTOWER 2.2 quantitative Real-time system. The reaction system consisted of 5μL 2×SYBR® Green Supermix, 1 μL cDNA, 0.5 μL forward primer, 0.5 μL reverse primer and 3 μL ddH2O. The reaction program was conducted as follows: step 1, 95°C for 3 min; step 2, 95 °C for 10 s; step 3, 60 °C for 30 s and plate read; go to step2, 39 cycles; melt curve analysis (60 °C 95 °C, + 1°C/cycle, holding time 4 s). The 2 −∆∆CT method was used to determine the relative expression levels of target genes in each sample individually.
3. RESULTS
3.1. Gonadal development stage determination
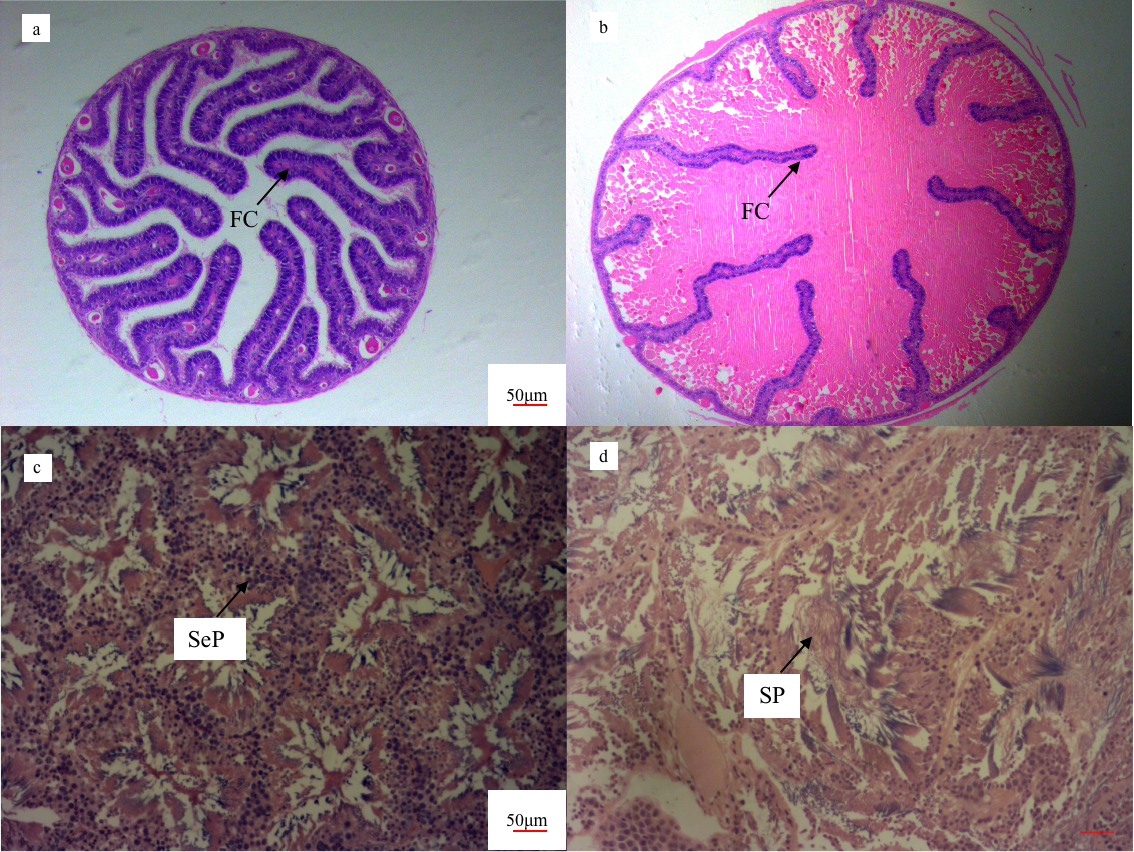
To verify the gonadal development stage of O. minor, we conducted histological analysis of the female and male gonads (Fig. 1), and distinguished stages based on description of O. tankahkeei.18 The samples showed sex differentiation obviously, the oocyte in the oravies were circle. In the immature female gonad (Fig. 1a), the cells were basophilic and stained dark blue. The follicular cells folded into the cells on a large scale, forming large and long folds and with less yolk material. The lipid particles could be observed obviously. Based on these, they were determined to the middle stage of oocyte development. In the mature female gonad (Fig. 1b), the cells are eosinophilic, stained light pink, less intrafollicular folding and more yolk, it is in the fourth stage of oocyte development. In the immature male octopus testis (Fig. 1c), a large number of secondary spermatocytes were distributed in the seminiferous lobules, and the head was stained dark blue, which is in the second stage of development. In the mature male octopus testis (Fig. 1d), a small number of spermatocytes and sperm show irregular hollow structure in the seminiferous lobules, which is in the fifth stage of development.

3.2. Overview of RNA-Seq results
Twelve libraries with 3 replicates each group were successfully constructed and sequenced. While the MM3 sample was eliminated for the high correlation coefficient (>0.8) with immature male group, after tested by correlation coefficients of differential expressed genes (Fig. S1). A total of 263,749,727 clean reads were obtained from the remaining 11 libraries, and 77.2%–90.59% reads of clean data were effectively matched to the genome of O. minor (Table S2). The percentage of uniquely mapped reads ranged from 69.11% to 84.55%. Quality assessment of the sequencing data showed that the Q30 of each sample was 93.9%–96.54, and the Q20 was 98.19%–98.98%. These data indicated that the sequencing quality can be effectively used for subsequent transcriptome analysis (Table S3). The sequences with too short peptide chains (less than 50 amino acid residues) or containing only a single exon were filtered out, and a total of 21785 new genes were discovered.
3.3. Differential Expression Analysis
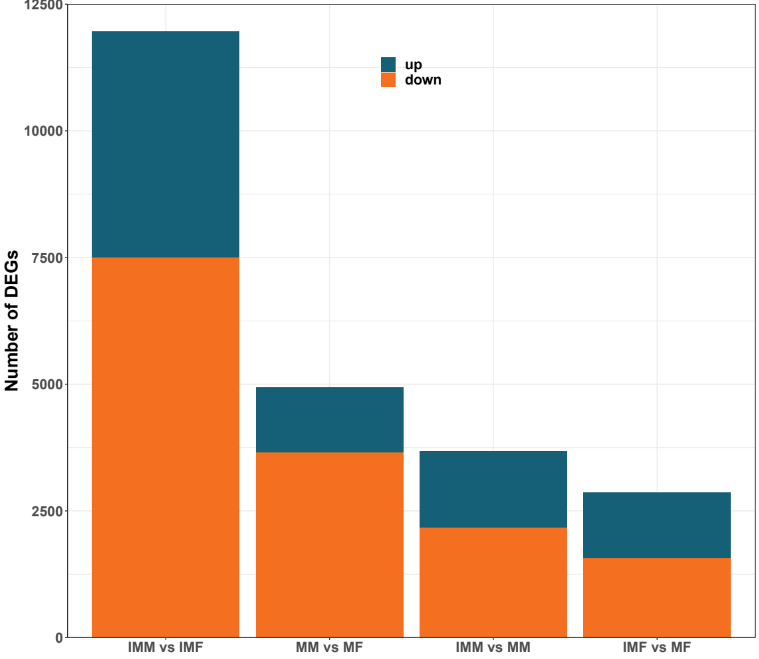
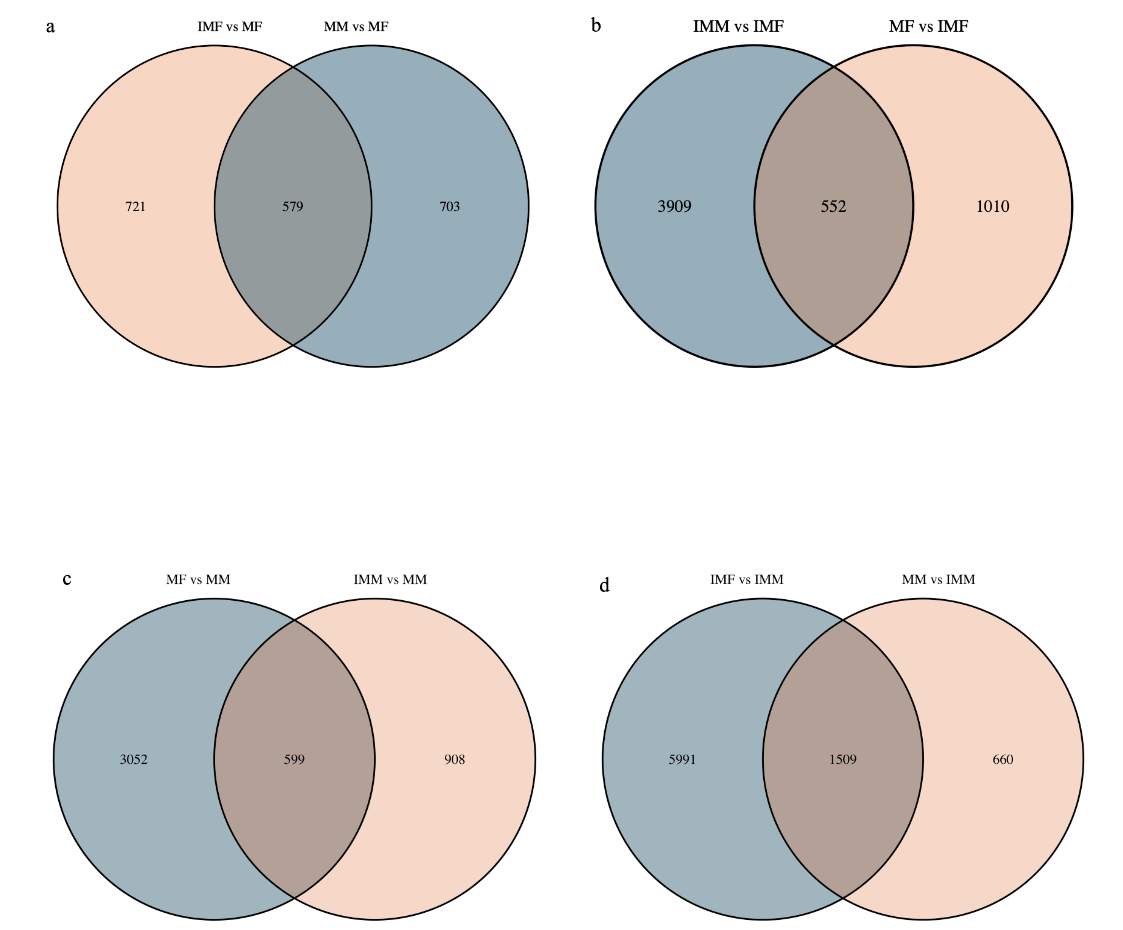
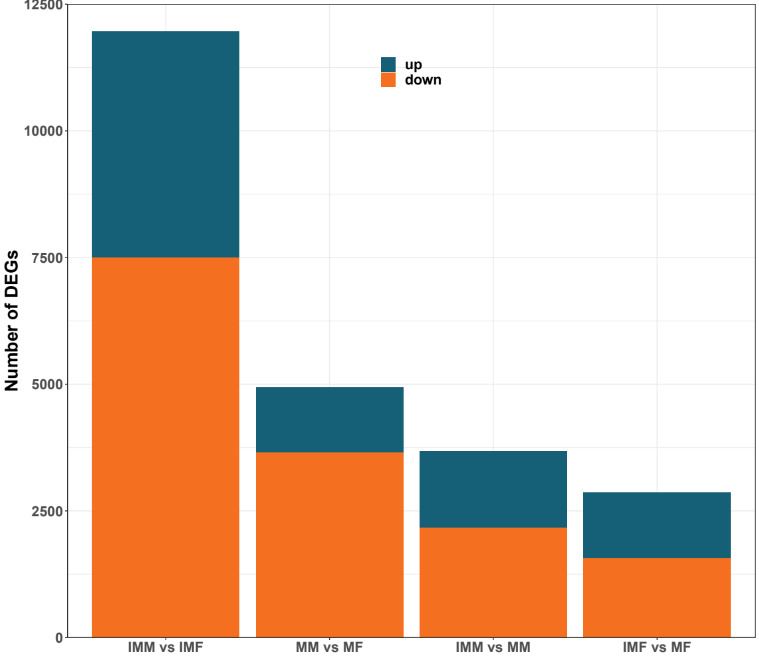
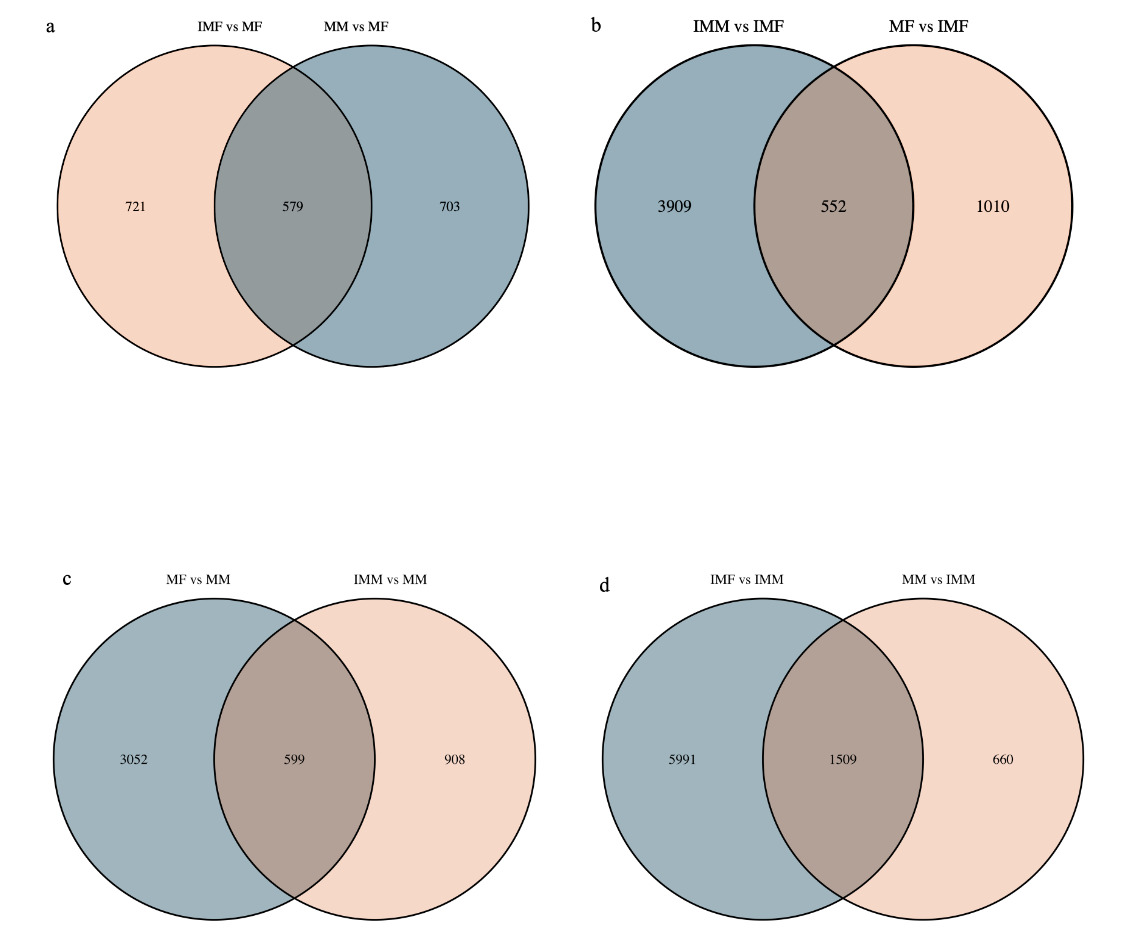
In the comparison of IMM vs IMF, 4461 DEGs were up-regulated, and 7500 DEGs were down-regulated. In the comparison of MM vs MF, 1281 DEGs were up-regulated, and 3651 DEGs were down-regulated. In the comparison of IMM vs MM, 1506 DEGs were up-regulated, and 2168 DEGs were down-regulated. In the comparison of IMF vs MF 1300 DEGs were up-regulated, and 1561 DEGs were down-regulated (Fig. 2). A Venn diagram analysis was carried out based on these DEGs. In the MM vs MF and IMF vs MF up-regulated DEGs comparison, we selected the rest of the MM vs MF group without the intersection as the female-regulated genes in the mature female octopus, and a total of 703 DEGs were revealed. At the same time, 3903, 3052 and 5991 DEGs were revealed as the sex-related genes in the immature female, mature male and immature male octopus respectively based on this method (Fig. 3). Among these DEGs, several genes of interesting were obtained. For example, folate receptor 1 protein (FOLR1), forkhead box protein L2 (Foxl2), ribosomal protein S9 (RPS9), ribosomal protein large 35a (RPL35a), 40S ribosomal protein small 17 (40sRPS17), ribosomal protein large 17 (RPL17) and ribosomal protein large (RPL24) were obtained as the candidate female-regulated genes. Meanwhile, Testis-specific serine/ threonine-protein kinase 2-like (TSSKs), neuropeptide Y receptor type 5 isoform X3 (NPYR5s), fem-1 protein C (Fem1C), neuropeptide S receptor (NPSR), transcription factor Sox8 (Sox8), tripartite motif-containing protein 2 (TRIM2) and bone morphogenetic protein-6 (BMP-6) were obtained as the candidate male-related genes.
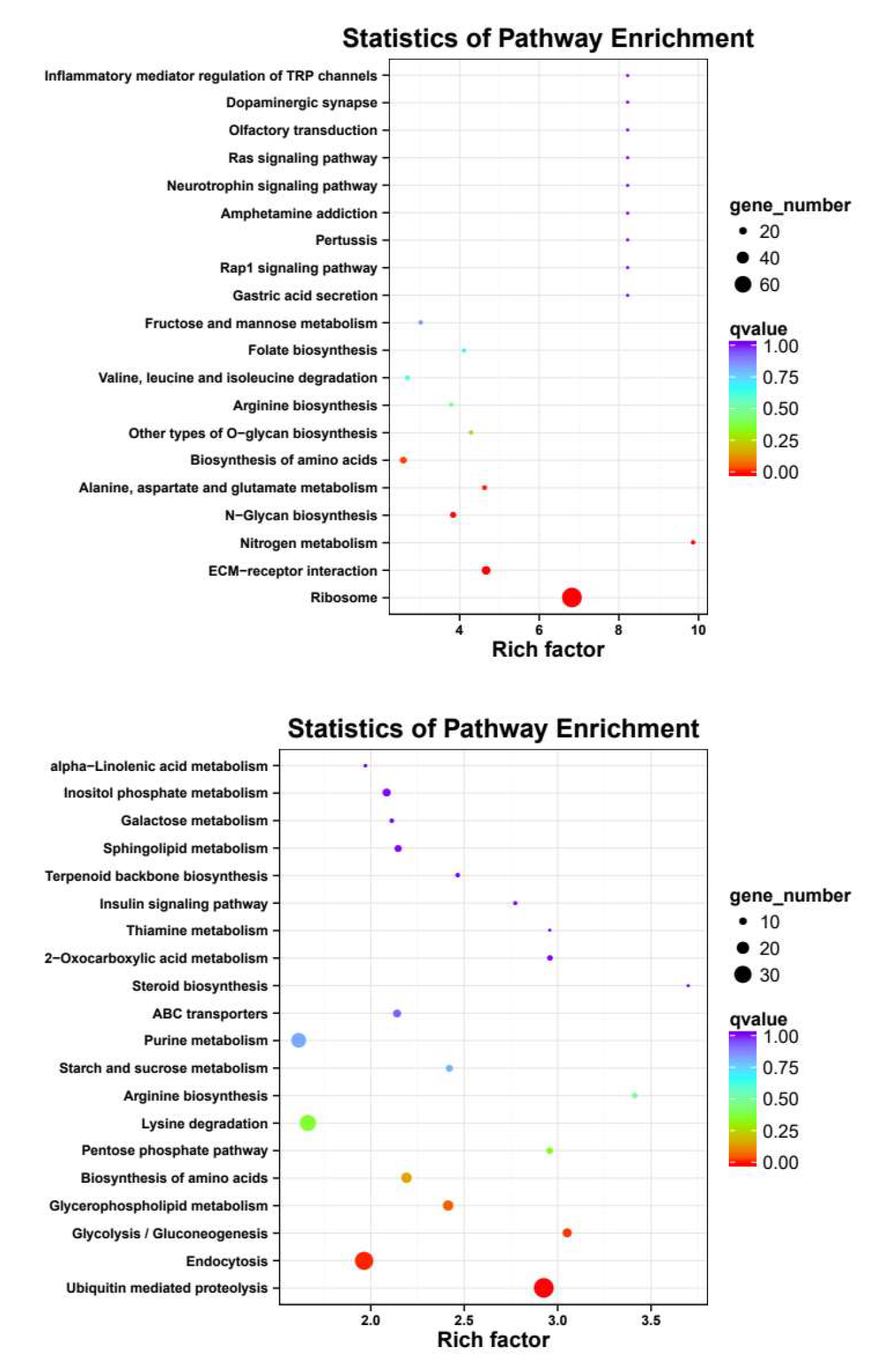
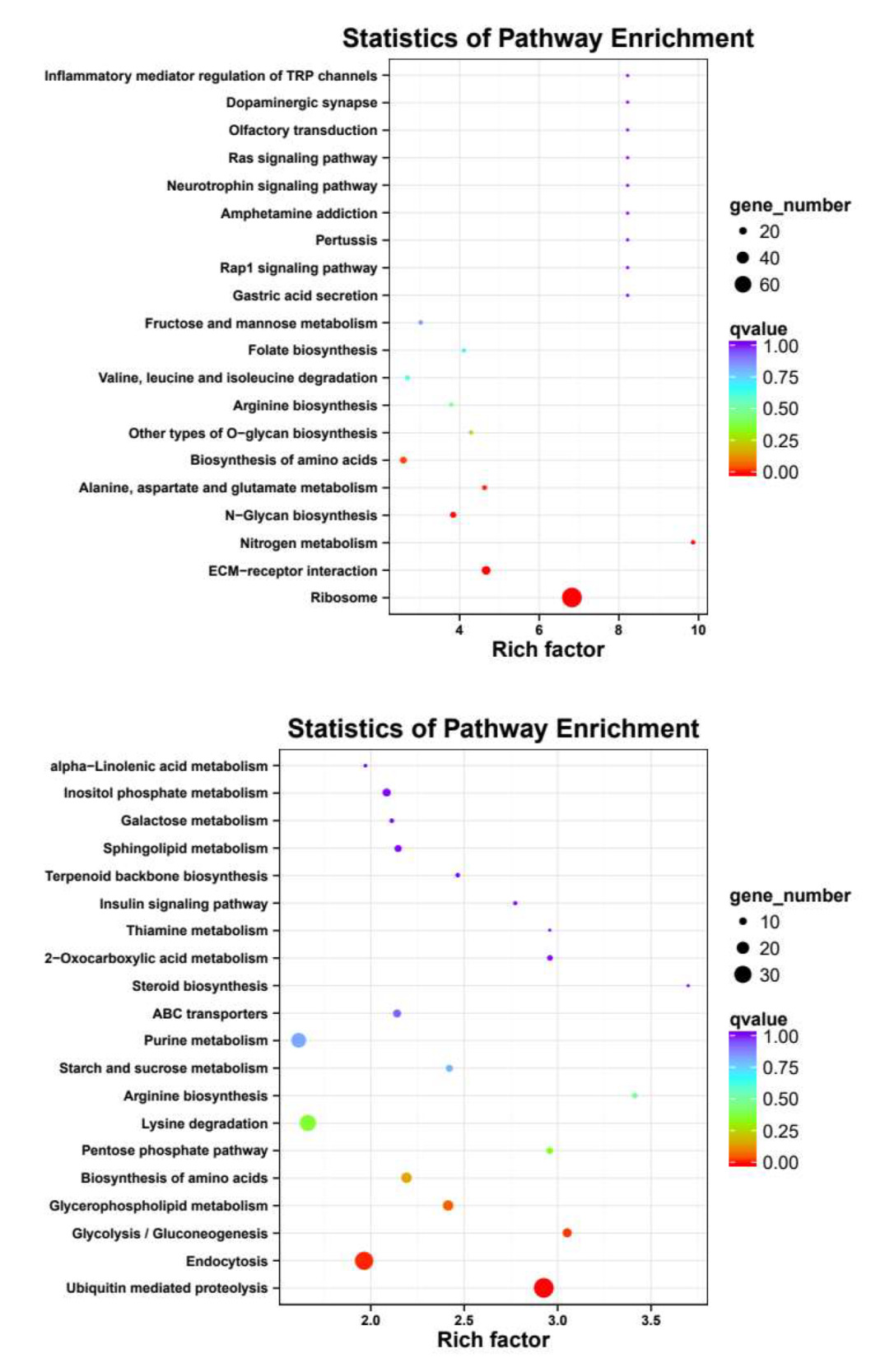
3.4. GO and KEGG enrichment analysis
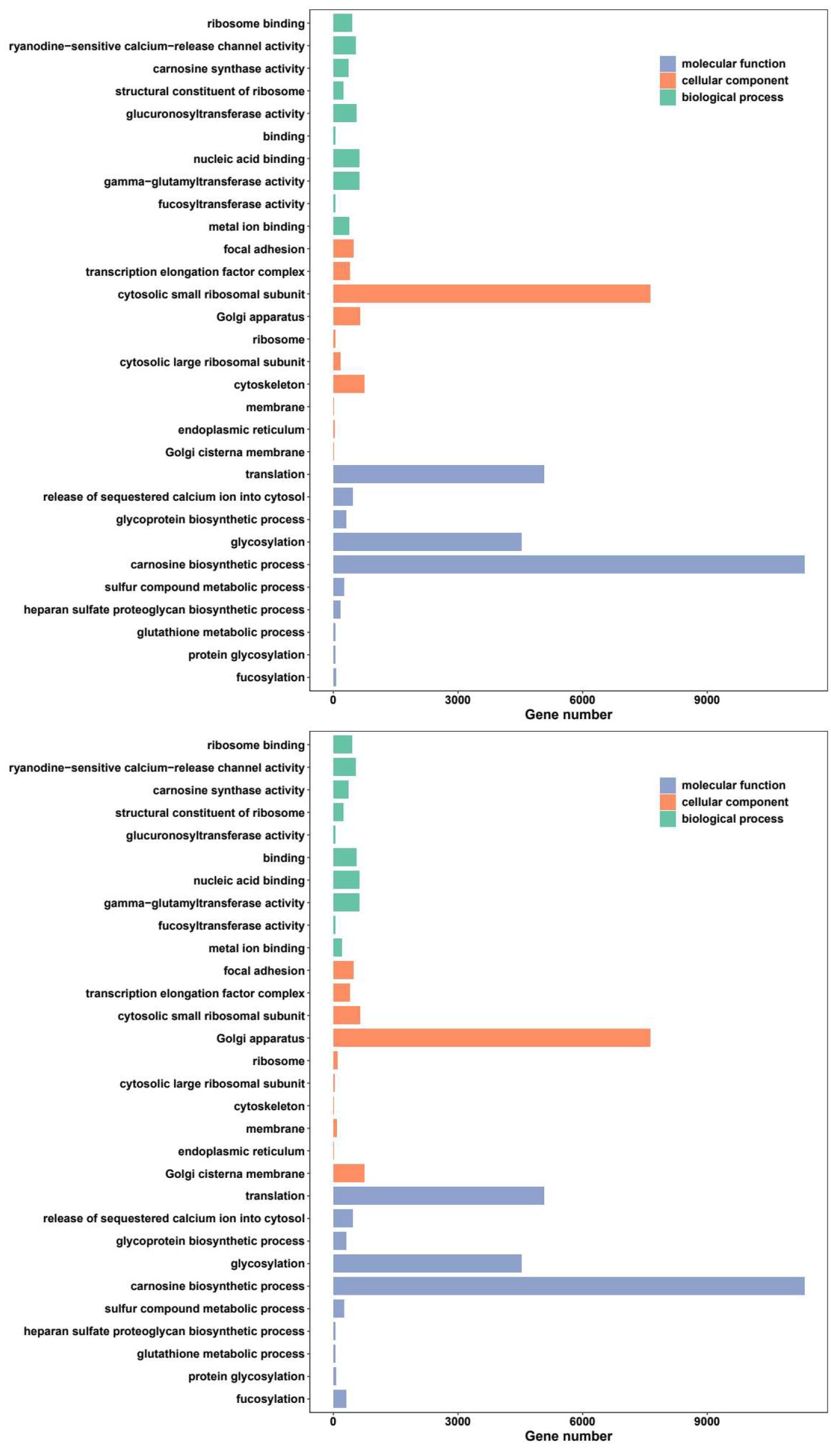
GO and KEGG enrichment analysis were employed based on the selected DEGs, respectively. The DEGs were assigned to various GO terms and three main functional categories: biological process (BP), cellular component (CC) and molecular function (MF), and the top ten terms were contrasted in the three categories individually (Fig. 4). Among the candidate group of female-regulated DEGs, all the top ten terms including cellular process, metabolic process, single-organism process, biological regulation, localization, response to stimulus, signaling, cellular component organization or biogenesis, biological adhesion and developmental process were both annotated. Among the candidate group of male-regulated DEGs, the same top ten terms enriched in female-regulated DEGs were annotated.

Meanwhile, KEGG pathway analysis was performed with the same four groups of DEGs (Fig. 5), revealing Ribosome, ECM-receptor interaction, N-Glycan biosynthesis, Ribosome biogenesis in eukaryotes, Valine, leucine and isoleucine degradation and Fructose and mannose metabolism were enriched in both female-biased DEGs, and Ubiquitin mediated proteolysis, Endocytosis, Lysine degradation, Glycolysis/Gluconeogenesis, 2-Oxocarboxylic acid metabolism, Glycerophospholipid metabolism were enriched in both male-biased DEGs.
3.5. qRT-PCR validation
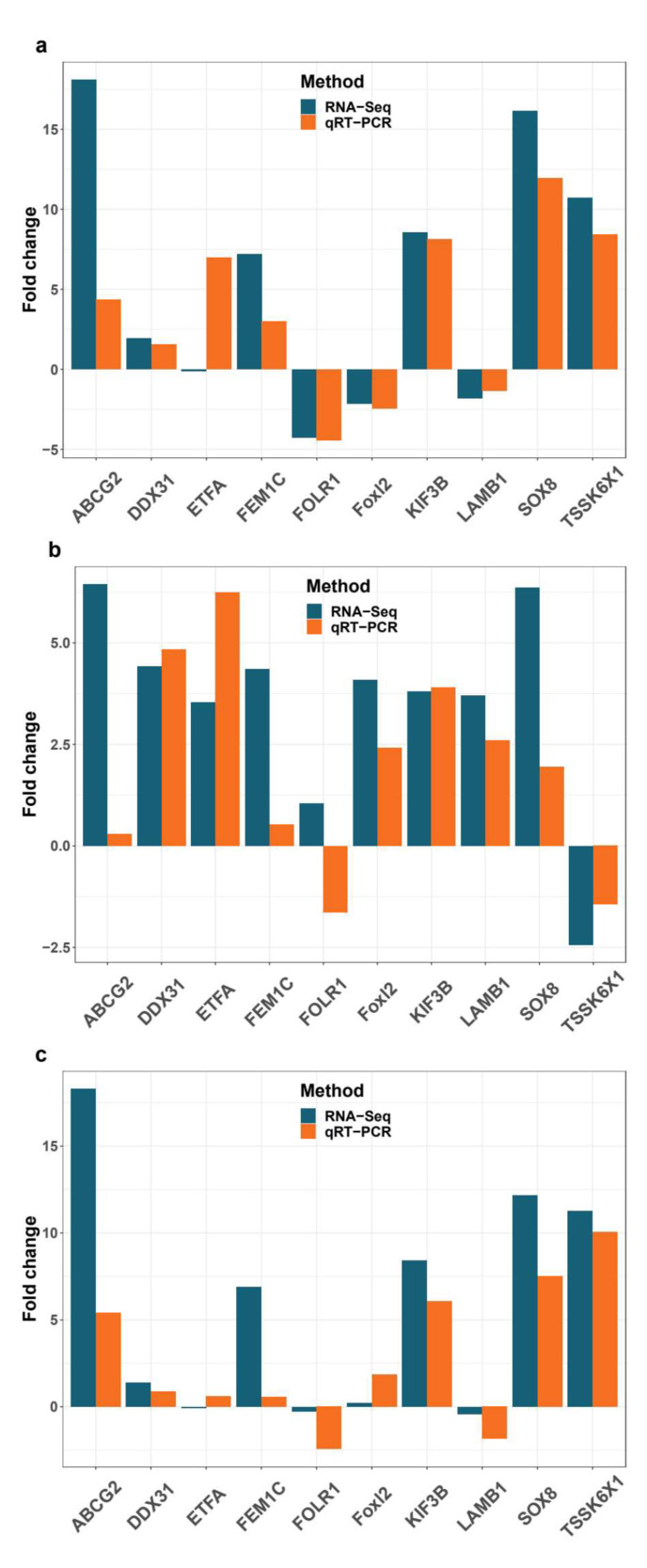
In order to validate the DEGs identified by RNA-seq, ten DEGs were tested using quantitative real-time PCR. Comparing the transcriptome data with the qRT-PCR results from eight selected DEGs, trends of quantitative qRT-PCR results were correlated with qRT-PCR results, and two DEGs, i.e. electron transfer flavoprotein subunit alpha (ETFA) and folate receptor 1protein (FOLR1), were not absolutely consistent with transcriptome results (Fig. 6). Overall, the qRT-PCR results indicated the reliability and accuracy of the RNA-Seq-based transcriptome expression analysis.
4. Discussion
Gonadal transcriptome sequence has been widely used to identify sex-determining genes in mollusks. Here, transcriptome analysis on the ovaries and testes of O. minor were conducted. Numerous genes and pathways that are potentially involved in sex-determining were identified, which provided important data for reproductive biology in O. minor.
4.1. Genes related to testis development
Of the DEGs that were identified between the male and female gonadal transcritpomes in O. minor, male-biased genes were selected. Several well documented and important genes are obtained, including Fem1C and Sox8, which were highly expressed in the testis compared to the expression level in the ovary. Those genes were considered to be related to the development of the testis of O. minor, including Sox8 and Fem-1.
The Sox genes encode a group of proteins characterized by the existence of a sex-determining region on Y chromosome (SRY) box,19 and are classified as 12 subfamilies according to the conservatism of protein and nucleic acid sequences. Among which, Sox8 is classified in the SoxE group, which contain a DNA-dependent dimerization domain, unique among Sox proteins. Previous researches showed that SoxE were highly expressed during mammalian testis development, and were considered to be essential for the proper function of Sertoli cells in neonatal and adult animals.20 In Paralichthys olivaceus, Sox8 exhibited obviously tissue-specific and was testis biased,21 implying the testis-related function of Sox8. In mice, Sox8 null mutations can cause Müllerian duct regression and lead to a decline in fertility at later stages of testis development.20 To the best of our knowledge, Sox8 was not found expressed differently in invertebrate gonads, while another member of SoxE, i.e. Sox9 was considered as the male-determining gene22 in all kinds of mollusks. In this study, Sox8 was found highly expressed in the testis tissue, which can imply that Sox8 may be the unique gene that related to male determination in O. minor, which need be tested in detail.
Fermentation-1 (Fem-1) was first reported functioning in a signal transduction pathway that controls sex determination in the Caenorhabditis elegans and was required for all aspects of testis development.23 Fem-1 gene family played a conserved role in all kinds of tissues, and was considered to participate in the basic biological processes widely.24 As one of the homologs of Fem-1 genes, Fem1C was considered as the gene associated to the spermatogenesis, and was not related to the determination of sex in vertebrates, for the Fem-1 ablation showed no adverse effects in the sexual development in mice.25 In previous researches in several marine organisms, Fem1C expressed significantly special higher in testis compared to other tissues, and was inferred to involved in sex determination and differentiation.26 In the present study, Fem1C has a significant increase in expression level in the testis, which can be inferred to the male-biased gene.
4.2. Genes related to ovary development
Ovary is the female reproductive system and responsible for the synthesis of estrogen and oogenesis. In this study, Foxl2 and RPL24 were identified as the most important female-biased genes in O. minor.
Foxl2 is a forkhead transcription factor, which functions for ovarian differentiation by estrogen production specifically in the female gonad either by binding directly to the promoter of Cytochrome P450 family 19 subfamily A member 1 (Cyp19a1) or by interacting with adrenal 4 binding protein/steroidogenic factor 1 (Ad4BP/SF-1) to enhance the Cyp19a1 transcription.27 Studies indicated that Foxl2 was the critical factor responsible for maintaining the mammalian ovarian phenotype by supposing genes involved in Sertoli cell-promoting from the early embryonic gonad throughout adult life.28 In fish, Foxl2 was implied to determine and maintain ovarian development via estrogen synthesis.29 Sexually dimorphic expression of Foxl2 occurs not only in O. minor but also in other mollusks.30,31 In Chlamys farreri, Foxl2 was considered playing a vital role in its oogenesis, as nucleus pulsation were observed in the ovaries after Foxl2 knockdown in the mRNA level.31 Meanwhile, in Crassostrea gigas, Cg-Foxl2 was implied to participate in male gonadic differentiation indirectly, based on down-regulation induced by its natural antisense transcript.32 In this study, Foxl2 was identified as an ovarian up-regulated gene. It implicated this gene defined as potential marker of female O. minor, while the function of Foxl2 in O. minor has yet to be established.
In this study, the expression level of RPL24 was much higher in testis than that in ovaries. Ribosomal protein genes (RPs) are indispensable in ribosome biogenesis and protein synthesis, and are highly conserved even between vertebrates and invertebrates.33 In previous studies, RPs were widely used as markers for phylogenetic studies and comparative genomics in teleosts.34 Over the past years, RPL24 was found expressed highly various between ovaries and testes in different organisms.33 In Marsupenaeus japonicus, the expression level of RPL24 in ovary is much higher than in testis, which was considered playing an important role in oogenesis.35 Moreover, RPL24 was considered having a positive correlation with the vitellogenesis and the oocyte maturation, for the ovarian histological observation showed the ovarian development was delayed by RPL24 gene silencing.36 In the present study, not only RPL24 was the testis-up-expression gene, but also the other RPs, such as RPS9, RPL35a, 40sRPS17, RPL17, showed the same trends, which may confirm RPs play a vital role in the male-determination in O. minor.
4.3. Candidate pathway in sex-determination
Of the KEGG pathways enriched by sex determining genes screened by Venn, ECM-receptor interaction pathway is the most potential sex marker for the O. minor. The extracellular matrix (ECM) supports many cellular processes, including adhesion, migration, proliferation, and survival,37 which occur in large numbers during ovarian development. Moreover, ECM regulates the processes that are necessary for follicle growth and oogenesis, such as the communication between the oocyte and companion follicle cells 38 and the synthesis of steroid hormone.39 In the present studies, ECM receptor interaction were inferred to play an important role in ovarian development in mice37 and some other mammals.40,41 In the study on mollusks, this pathway is also implicated in ovarian function in Crassostrea hongkongensis.42 In conjunction with these previous studies, our results suggested that the ECM-receptor interaction pathway was associated with the female-determination and maintenance in O. minor.
5. Conclusion
This study screened potential sex determining genes and signal pathways through gonadal transcriptome analysis of male and female O. minor at different developmental stages. Sox8 and Fem-1 were identified as the male-biased genes, and Foxl2 and RPL24 were identified as the female-biased genes, and ECM-receptor interaction pathway was the most potential sex marker, which provided a reference for explaining the molecular mechanism of sex determination in O. minor. Meanwhile, the biological function of DEGs screened by RNA-Seq in O. minor were not clear, and the sex-determination hub genes were not found. Therefore, it’s necessary to explore detailed molecular mechanism of sex determination of these genes and the hub genes, so as to establish a set of O. minor sex determination system, bringing a breakthrough in resource enhancement and aquaculture industry of O. minor.
Acknowledgements
This study was supported by Shandong Key Research and Development Program (2019GHY112022), China Agriculture Research System (CARS-49), Yantai Comprehensive Test Station of Shandong Shellfish Industry Technology System (SDAIT-14-11).
Author contributions
Jiabei He analysed data and writed the paper; He Wang designed study and collected the samples; Chao Liu collected the samples and analysed data; Siqing Chen analysed data; Qiang Zhao revised paper; Jianlong Ge designed study and revised paper; Yongsheng Liu designed study and drafted paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Data availability statement
The raw data were submitted to the NCBI (accession number: SAMN27553764–SAMN27553775).