Introduction
Tilapia is an important aquaculture species due to its wide acceptance, environmental adaptability, and strong disease resistance. Pond culture is a crucial approach for freshwater aquaculture and ensures high-quality protein availability for human consumption. Recently, rice-fish symbiosis has become prevalent. This approach leverages the mutually beneficial symbiotic relationship between rice and fish to enhance farming’s ecological conditions. Rice-fish symbiosis plays a vital role in improving aquaculture’s economic and social benefits. Meanwhile, the rice-fish symbiosis could improve the growth performance of loaches, enhance the abundance and diversity of intestinal microbiota, enhance immunity and disease resistance, and regulate liver metabolism.1 Studies have also shown differences in the diversity of the gut microbiota between different farmed fish in the same paddy field and at different growth stages of the same fish.2 Nie et al. showed that gut microbiota affects the growth of rice carp by regulating metabolism.3 In general, rice-fish symbiosis, as an ecological farming model, can improve fish’s muscle nutrients and intestinal flora diversity.
The intestinal microbiota’s composition is crucial in maintaining the host’s physiological and metabolic homeostasis, and it is closely related to the host’s nervous, endocrine, and immune system development and function.4–6 There are 1×103~1×104 types of microorganisms in the human gastrointestinal tract, which is ten times the number of human cells. As a result, intestinal microorganisms are often referred to as “forgotten organs”.7 Intestinal microorganisms are divided into exogenous and native groups related to gastrointestinal digestion. However, exogenous microorganisms are temporary and free-floating, while native microorganisms are the core community that colonizes the digestive tract’s surface and prevents foreign organisms from colonizing the intestinal surface.8,9 Intestinal microorganisms include fungi, yeasts, viruses, bacteria, and archaea, among which bacteria are the primary intestinal microorganism flora. Due to the significant impact of intestinal bacteria on the host’s health, research on the diversity, function, and mechanism of action of intestinal bacteria has become a hot topic in biological research in recent years.
The composition of animals’ intestinal microbiota is closely linked to various factors, including environmental factors, physiological state, and life developmental stage.10,11 As aquatic animals reside in a water environment and possess a short digestive tract, their intestinal microbiota’s relative abundance is susceptible to external environmental changes. This susceptibility can adversely affect farmed animals’ health and their intestinal microbial composition.12,13 The growth and development of fish are significantly influenced by intestinal flora, which aids in nutrient digestion and absorption while regulating the host’s defense function, promoting healthy fish development. To understand the effects of different farming methods on the intestinal flora composition and muscle nutrient profile of tilapia, this study utilized 16SrRNA high-throughput sequencing technology. The results provide a theoretical basis for understanding the intestinal micro-physiology of tilapia under different farming methods.
Materials and Methods
Experimental design and sampling
The samples were divided into two groups:
R group (Rice-fish symbiosis group): the rice field size was 25 m *40 m (fish ditch: 25 m *4 m *1.5 m), 900 tilapias (15.26±0.98g/ tail) were stocked in the rice field and fed 31% protein compound diet for 4 months. 9 tilapias were randomly collected, and 4 of them were tested for muscle nutrients.
T group (Tank field group): 900 tilapias (15.26±0.98 g/ tail) were put into each pond (20 m *10 m *1.5 m) and fed 31% protein compound diet for 4 months. 9 tilapias were randomly collected, and 4 of them were tested for muscle nutrients.
Amino acid and fatty acid contents in muscle
The muscle nutrient composition was evaluated by detecting the amino acid composition and fatty acid content according to previous studies.14 Briefly, the muscle sample was ground into a powder with liquid nitrogen; 40 mg of freeze-dried muscle powder and 10 mL of 6M hydrochloric acid were placed in a 50 mL ampoule, which was then placed in a constant temperature drying oven to hydrolyze for 24 h. The hydrolysate was diluted with deionized water, transferred to the rotary evaporator flask, and evaporated to dryness (60°C). The evaporating product was washed with 0.02 M hydrochloric acid and transferred to another volumetric flask. A 3 mL hydrolysate sample and standard amino acid solution were respectively filtered into an automated sample injection bottle, and the amino acid content was determined by the automatic amino acid analyzer (LA8080, Hitachi, Japan).
The fatty acid content was determined according to the following steps: 0.5 g of freeze-dried muscle powder and 4 mL mixture of benzene and petroleum ether (volume ratio 1:1) were put into a 10 mL centrifuge tube to extract for 24 h. 4 mL potassium hydroxide-methanol solution (0.4 M) was added into the centrifuge tube for methyl esterification, and the mixture was vortexed for 3 min and stood for 30 min. The mixture was diluted with deionized water and the upper layer solution was extracted. 200 μL upper layer solution and 800 μL hexane were mixed and filtered with 0.22 μm filter membrane. The gas chromatograph GC 2010 (A09056) was used to determine the fatty acid content.
16S RNA gene sequencing for the intestinal microbiota analysis
According to the manufacturer’s instructions, DNA from different samples was extracted using the E.Z.N.A. ®Stool DNA Kit (D4015, Omega, Inc., USA). The reagent, designed to uncover DNA from trace amounts of sample, is effective for the preparation of DNA of most bacteria. Nuclear-free water was used for blank. The total DNA was eluted in 50 μL of Elution buffer and stored at -80 °C until measurement in the PCR by LC-Bio Technology Co., Ltd, Hang Zhou, Zhejiang Province, China. The 5’ ends of the primers were tagged with specific barcodes per sample and sequencing universal primers. PCR amplification was performed in a total volume of 25 μL reaction mixture containing 25 ng of template DNA, 12.5 μL PCR Premix, 2.5 μL of each primer, and PCR-grade water to adjust the volume. The PCR conditions to amplify the prokaryotic 16S fragments consisted of an initial denaturation at 98°C for 30 seconds, 32 cycles of denaturation at 98 °C for 10 seconds, annealing at 54 °C for 30 seconds, and extension at 72 °C for 45 seconds, and then final extension at 72 °C for 10 minutes. The PCR products were confirmed with 2% agarose gel electrophoresis. Ultrapure water, instead of a sample solution, was used throughout the DNA extraction process to exclude the possibility of false-positive PCR results as a negative control. The PCR products were purified by AMPure XT beads (Beckman Coulter Genomics, Danvers, MA, USA) and quantified by Qubit (Invitrogen, USA). The amplicon pools were prepared for sequencing, and the size and quantity of the amplicon library were assessed on Agilent 2100 Bioanalyzer (Agilent, USA) and with the Library Quantification Kit for Illumina (Kapa Biosciences, Woburn, MA, USA), respectively. The libraries were sequenced on the NovaSeq PE250 platform.
Samples were sequenced on an Illumina NovaSeq platform according to the manufacturer’s recommendations provided by LC-Bio. Paired-end reads were assigned to samples based on their unique barcode and truncated by cutting off the barcode and primer sequence. Paired-end reads were merged using FLASH. Quality filtering on the raw reads was performed under specific filtering conditions to obtain high-quality clean tags according to the fqtrim (v0.94). Chimeric sequences were filtered using Vsearch software (v2.3.4). After dereplication using DADA2, we obtained the feature table and feature sequence. Alpha diversity and beta diversity were randomly calculated by normalizing them to the same sequences. Then, according to the SILVA (release 132) classifier, feature abundance was normalized using the relative abundance of each sample. Alpha diversity was applied in analyzing the complexity of species diversity for a sample through 5 indices, including Shannon, Chao1, Observed OTUs, Goods coverage, and Simpson, and all these indices in our samples were calculated with QIIME2. Beta diversity was calculated by QIIME2; the graphs were drawn by the R package. Blast was used for sequence alignment, and the feature sequences were annotated with SILVA database for each representative sequence. Other diagrams were implemented using the R package (v3.5.2). The corresponding graphs and heatmaps were drawn using the OmicStudio tools at https://www.omicstudio.cn/tool.
Statistical analysis
A one-way analysis of variance (ANOVA) was used to evaluate significant differences between the between-groups using SPSS 25.0 (Chicago, IL, United States). p< 0.05 was set as the significance threshold. Spearman’s correlation analysis was conducted to analyze the relationship between microbiota and muscle nutrition using data from significantly different genus-level microbiota and differential muscle nutrition.15 Using the default clustering method, a scaled heatmap was constructed for the correlation matrix.
Results
The growth performance of tilapia in two groups
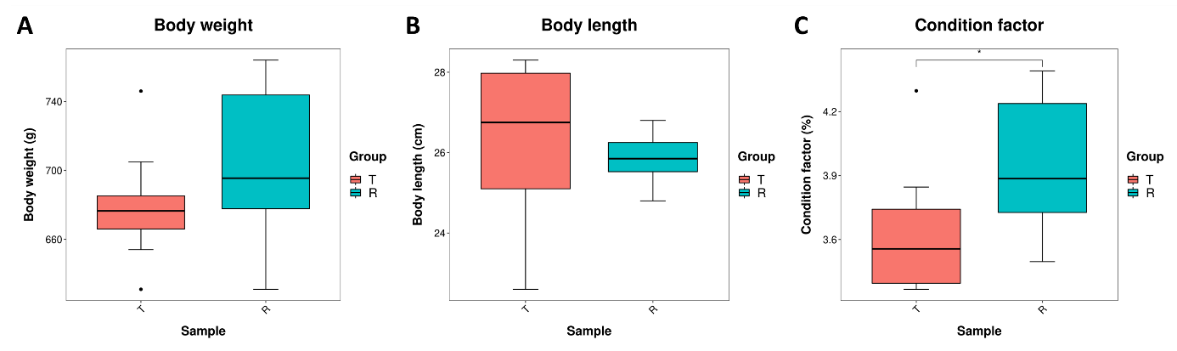
Both groups of tilapias were fed the same amount of feed. Studies have shown that the degree of CF (Condition factor) is closely related to the body fat content of fish. To observe the changes in the body size of tilapia, body weight, and body length were measured separately, and the corresponding fullness was calculated. Figure 1 showed that the weight of tilapia in the R group was slightly higher than that in the T group (Figure 1A). The body length of the T group was slightly higher than that in the R group (Figure 1B), but there was no significant difference in tilapia weight or body length between the two groups. However, the Condition factor of tilapia in the R group was significantly higher than that in group T (Figure 1C) (p<0.05).

Amino acid and fatty acid content in muscle
The content and composition of amino acids and fatty acids are important indicators for evaluating the muscle quality of fish.16 To explore the changes in the nutrient composition of tilapia muscle under the two modes, we analyzed the composition and content of amino acids and fatty acids in tilapia muscle. In this study, a total of 17 free amino acids were detected from tilapia muscle (Table 1). The highest content was Glu (Glutamic acid), and the lowest was Cys (Cysteine). The contents of Val (Valine) and Phe (Phenylalanine) in tilapia in the R group were significantly higher than those in the T group. Meanwhile, the contents of TAA (total amino acid), TEAA (total essential amino acid), and TUAA (total umami amino acid) in the R group were higher and more diverse than those in the T group. In addition, 12 fatty acids were detected in tilapia muscle, including 5 saturated fatty acids and 7 unsaturated fatty acids (Table 2). It is worth noting that the DHA (Docosahexaenoic acid) content of tilapia muscle in the R group was significantly higher than that in the T group (p<0.05).
Operational taxonomic units (OTUs) and intestinal microbiota diversity analysis
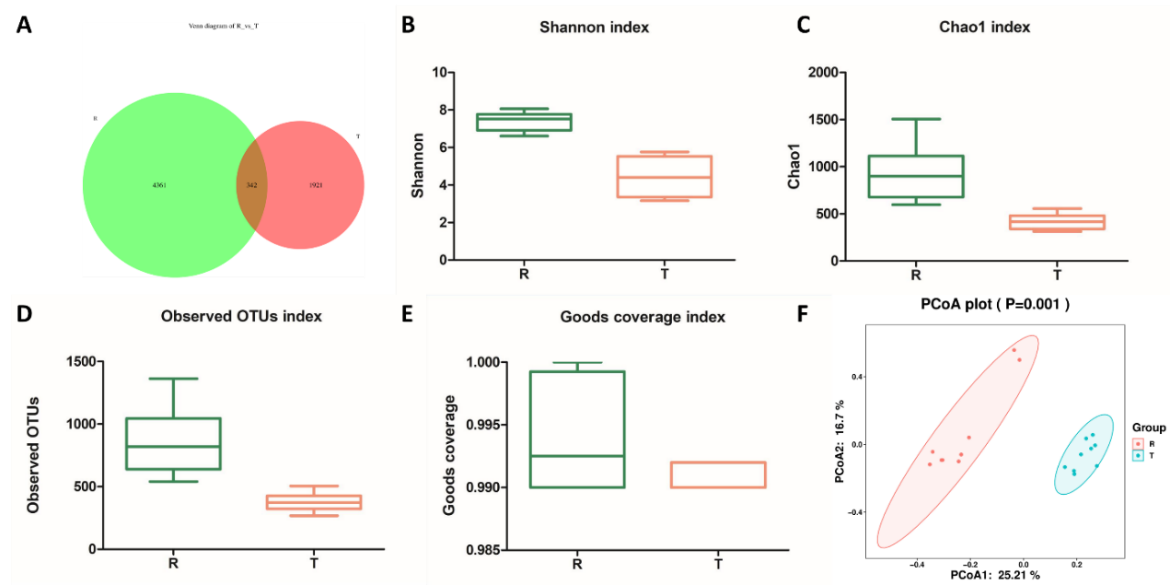
In order to explore the structure of intestinal flora in the process of pond culture and rice field culture, the 16S rDNA technology was used to analyze the intestinal microbiota of tilapia, and the sequence similarity greater than or equal to 97% was classified as an OUT unit by ASVs. To observe the number of common and unique OTUs of each group, the number of common features of each group is calculated and presented through a Venn graph. The number of intestinal microbiotas OUT in the R group was 4361, which was higher than that in the T group (1921), and the same OUT in the two groups was 342 (Figure 2A). The diversity analysis results showed that the Shannon index, Chao1 index, Observed OTUs index and Goods Coverage index of the R group were significantly higher than those of the T group (p<0.05, Figure 2B-E). Meanwhile, the results of Principal coordinate analysis (PCoA) showed that the samples were clustered together between the groups. The distance between the R and T groups was far away (Figure. 2F). The results indicated that the intestinal flora structure of tilapia was similar in each group. The two groups had differences in the intestinal flora composition of tilapia.

The composition and clustering analysis of intestinal flora in the two groups
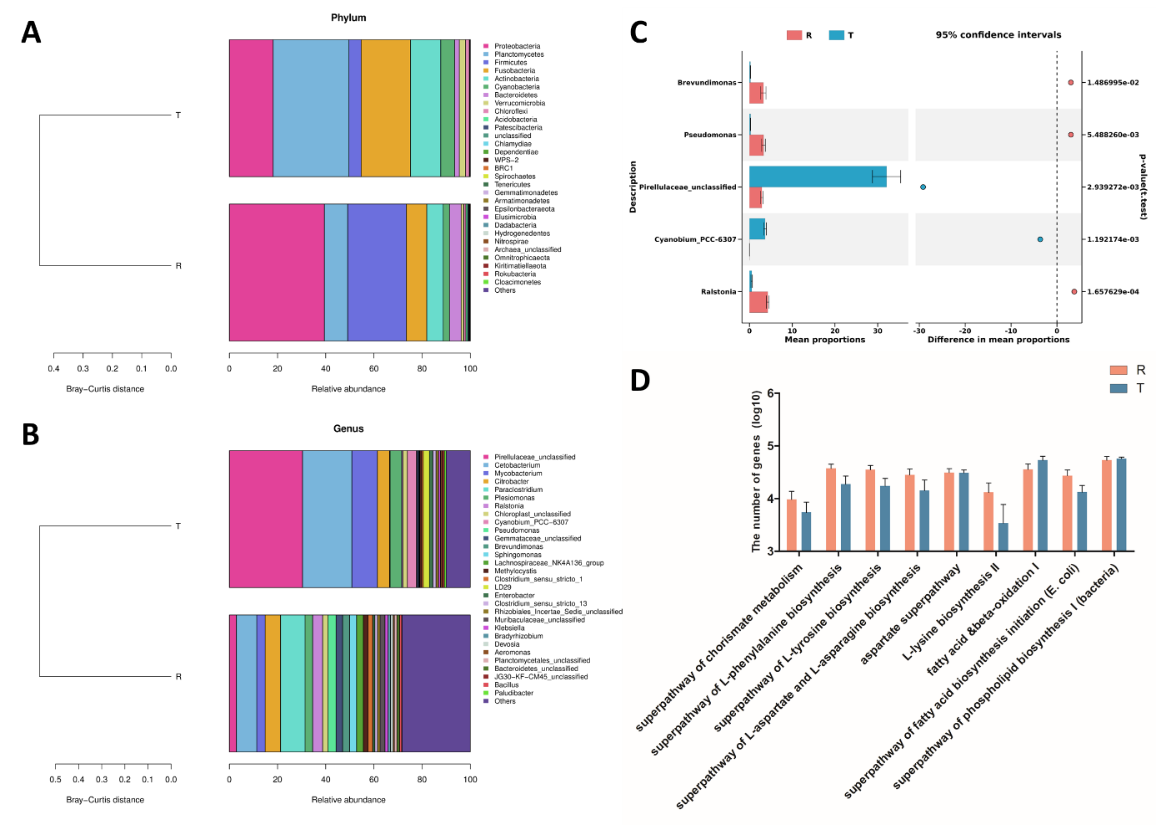
The composition of intestinal flora in two groups was further analyzed, and the sequences were identified. At the phylum level, the top 10 bacteria in relative abundance were Proteobacteria, Planctomycetes, Firmicutes, Fusobacteria, Actinobacteria, Cyanobacteria, Bacteroidetes, Verrucomicrobia, Chloroflexi, and Acidobacteria (Figure 3A). At the genus level, the top 10 bacteria were: Pirellulaceae_unclassified, Cetobacterium, Mycobacterium, Citrobacter, Paraclostridium, Plesiomonas, Ralstonia, Chloroplast_unclassified, Cyanobium_ PCC-6307 and Pseudomonas (Figure 3B). In order to explore the differences in the composition of intestinal flora between the two groups of tilapias, the differential microbiota was analyzed at the genus level. The results showed that the abundance of Brevundimonas, Pseudomonas, and Ralstonia in the R group was significantly higher than those in the T group (p<0.05), but Pirellulaceae_unclassified and Cyanobium_PCC-6307 were opposite (Figure 3C). PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) is a software that predicts functional abundance based on marker gene sequences. Based on the 16S rDNA gene sequencing data and KEGG (Kyoto Encyclopedia of Genes and genomes) database, 10 sub-functions related to muscle nutrition were found in two groups of tilapia intestinal bacteria at the secondary functional level (Figure 3D). Among them, the predicted functional gene numbers of superpathway of chorismate metabolism, superpathway of L-phenylalanine biosynthesis, superpathway of L-tyrosine biosynthesis, superpathway of L-aspartate and L-asparagine biosynthesis, aspartate superpathway, L-lysine biosynthesis II & beta-alanine biosynthesis II, fatty acid & beta -oxidation I and superpathway of fatty acid biosynthesis initiation (E. coli) in group R were higher than those in group T.

Correlation analysis of 16S sequencing and metabolomics
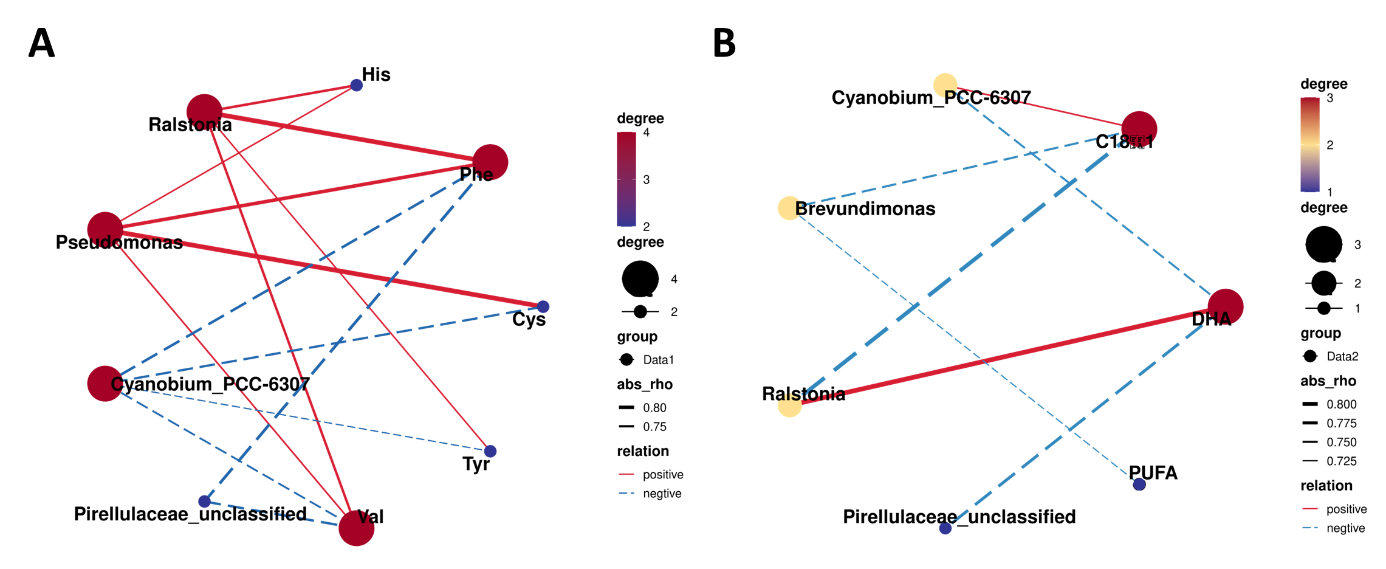
Spearman analysis was performed using intestinal differential bacteria (Top 5) of tilapia with differential muscle amino acids and fatty acids to explore further the correlation between intestinal flora and muscle nutrients of tilapia in the T group and R group. The results showed that the abundance of Ralstonia was positively correlated with the contents of His (Histamine), Phe (Phenylalanine), Tyr (Tyrosine), Val (Valine), and DHA (Docosahexaenoic acid), respectively. The abundance of Pseudomonas was positively correlated with the contents of His, Phe, Cys (Cysteine), and Val, respectively (Figure 4). While the abundance of Pirellulaceae_unclassified was negatively correlated with the contents of Phe, Val, and DHA, respectively. Furthermore, the abundance of Cyanobium_PCC-6307 was negatively correlated with Phe, Cys, Tyr, Val, and DHA contents, respectively.
_and_muscle_nutrition_in_tilapia.png)
Discussion
High-density pond culture can negatively affect the quality of fish muscle.17 The rice-fish symbiosis is an eco-breeding pattern. Studies have confirmed that rice-fish symbiosis could improve the quality of fish muscle. Meanwhile, the growth cycle of tilapia is almost the same as that of rice, and tilapia is also a suitable farmed fish for rice-fish symbiosis.18 In this study, we analyzed the composition of amino acids and fatty acids in the tilapia muscle from rice-fish symbiosis and pond culture. We explored the structure of the intestinal flora in each group. The results showed that the R group could increase the contents of Val, Phe, and DHA in the tilapia muscle compared to the T group. Additionally, the intestinal flora composition of tilapia differed between the two groups. The Shannon index, Chao1 index, Observed OTUs index, and Goods Coverage index of the R group were significantly higher than those of the T group (p< 0.05). Meanwhile, the abundance of Brevundimonas, Pseudomonas, and Ralstonia in the R group was significantly higher than that in the T group (p< 0.05), but the abundance of Pirellulaceae_unclassified and Cyanobium_PCC-6307 were opposite. It has been shown that Brevundimonas plays a positive role as beneficial gut bacteria in enhancing intestinal immunity and maintaining gut health in fish.19 Pseudomonas may help fish digest proteins and most secrete lipases to break down lipids for their own metabolic needs.20,21 At the secondary functional level, 10 sub-functions related to muscle nutrition were found in the gut microbes of the two groups of tilapias. Correlation analysis showed that the abundance of Pirellulaceae_unclassified was positively correlated with the opposite. At the secondary functional level, 10 sub-functions related to muscle nutrition were found in the gut microbes of the two groups of tilapias. Correlation analysis showed that the abundance of Pirellulaceae_unclassified was positively correlated with the contents of C18:1, DHA, Cys, and Phe, respectively. The abundance of Ralstonia was positively correlated with PUFA and Phe content, respectively, and the abundance of Cyanobium_PCC-6307 was positively correlated with the content of Phe and Cys, respectively.
The amino acid composition of muscle is related to taste. Studies have shown that the Phe and Val can enhance umami.22 In this study, the contents of Phe and Val of tilapia in the R group were higher than those in the T group, indicating that the rice-fish symbiosis could improve the muscle taste of tilapia. Studies have shown that DHA can enhance memory.23 In this study, the content of DHA in the muscle of tilapia cultured in the R group was higher than that in the T group, indicating that the rice-fish symbiosis can improve the nutritional quality of the tilapia muscle. In addition, the two groups had no significant difference in body weight and length. Still, CF in the R group was higher than that in the T group, which may be related to the fact that the SFA content of tilapia muscle in the R group was higher than that in T group. In general, the rice-fish symbiosis could increase the content of umami amino acids and DHA in tilapia muscle, improving the muscle quality of tilapia.
The fish’s intestinal system is vital for digestion, and research suggests that its microbiota plays an important role in nutrient metabolism and immune function regulation.24–26 Our research indicated that there was much more gut microbial OUT in the R group tilapia than in the T group. Meanwhile, the Shannon, Chao1, Observed OTUs and Goods coverage indexes of tilapia in the R group were higher than those in the T group. Combined with the result of beta diversity analysis, the intestinal flora diversity of tilapia in the R group was higher than that in the T group, which is consistent with the previous report.18 Further analysis of the intestinal flora composition of tilapia showed that the dominant groups of intestinal flora in the two groups were Proteobacteria, Planctomycetes, Firmicutes, Fusobacteria, Actinobacteria, Cyanobacteria, and Bacteroidetes, which is consistent with the results of previous studies.18 In previous studies, Pseudomonas was reported to have beneficial functions such as improving plant nutrition, degrading toxic substances, producing antibiotics, and inducing systemic resistance. At the same time, Ralstonia is an important inflammatory factor in the intestine.27–32 The study further showed that the abundance of Pseudomonas and Ralstonia in the R group was significantly higher than in the T group (p<0.05). We speculate that the composition of tilapia intestinal flora in the rice-fish symbiosis mode is more conducive to fish health.
New research has suggested that changes in the intestinal flora of fish are correlated with changes in their muscle nutrition.33,34 Through functional prediction analysis of PICRUSt2, we found that the secondary functional layer of intestinal bacterial genes mainly included amino acid and fatty acid metabolism in the rice-fish symbiosis of tilapia. Our research showed that the superpathways of chorismate metabolism, the superpathways of L-phenylalanine biosynthesis, the superpathway of L-tyrosine biosynthesis, the superpathway of L-aspartate and L-asparagine biosynthesis, the aspartate superpathways, the L-lysine biosynthesis II, the β-alanine biosynthesis II, the fatty acid β-oxidation I, and the superpathways of fatty acid biosynthesis initiation (E. coli) in the gut of tilapia in the R group had a higher number of predicted genes than T group. Therefore, we speculated that under different farming modes, the composition and abundance of different intestinal flora exert their respective functional genes and affect the metabolic pathways of nutrients such as amino acids and fatty acids in tilapia, thus showing the difference in muscle quality between the two modes. In addition, the correlation analysis showed that the abundance of Ralstonia was positively correlated with the contents of Phe, Val, and DHA. Still, the abundance of Pirellulaceae_unclassified was negatively correlated with the contents of Phe, Val and DHA, respectively. At the same time, the abundance of Ralstonia in the R group was higher than that in the T group, but the abundance of Pirellulaceae_unclassified in the R group was lower than that in the T group. Combined with the muscle nutrition results, we hypothesized that adding Ralstonia may improve the muscle quality of tilapia, and the underlying mechanism needs further investigation. Currently, the correlation analysis between the two bacterial genera and the associated nutritional components is merely a surface-level analysis, lacking a more in-depth understanding of the underlying molecular mechanisms within the fish. As a result, our future research efforts will focus on delving deeper into the molecular mechanisms involved, which will provide a more comprehensive theoretical foundation for our understanding of the relationship between the intestinal microbiota structure and the nutritional components of fish.
In summary, we found that the rice-fish symbiosis model is superior to the traditional culture model. Improving ecosystem interactions and increasing the diversity of gut flora improves tilapia’s muscle quality and nutritional value. It is important to note that the current study has some shortcomings, and future studies can expand the scope of research subjects, metabolic pathways, and molecular mechanisms to validate further and support these findings. In conclusion, the rice-fish symbiosis model provides a theoretical basis for optimizing and improving aquaculture practices, a reliable solution for promoting the sustainable development of aquaculture, and a reference for other traditional agricultural farming practices.
Author contributions
Formal Analysis: Yuanming Zhu (Equal), Yan Ji (Equal), Xuan Zhou (Equal), Xianlin He (Equal), Xiaoshu Xue (Equal), Jiaqi Zhang (Equal), Hongyu Tang (Equal), Ya Zhou (Equal), Chi Zhang (Equal). Writing – original draft: Yuanming Zhu (Equal), Yan Ji (Equal), Ya Zhou (Equal). Validation: Yan Ji (Equal), Xuan Zhou (Equal), Ya Zhou (Equal). Funding acquisition: Hongyu Tang (Equal), Chi Zhang (Equal). Supervision: Ya Zhou (Equal), Chi Zhang (Equal). Writing – review & editing: Ya Zhou (Equal), Chi Zhang (Equal).
Acknowledgments
This work was supported by project of science and technology re search program of the Research and Innovation Initiatives of WHPU, grant number 2023Y36; the Chongqing Aquatic Science and Technology Innovation Key Project, grant number CQFTIU2024-11; The start-up fund of WHPU, grant number 202410496041; Research Fund for the Doctoral Program of WHPU, grant number 2024RZ033.
Conflicts of Interest
The authors declared that they have no conflicts of interest to this work.
Ethics approval
All procedures and investigations were reviewed and approved by Wuhan Polytechnic University and were performed in accordance with the Guiding Principles for the care and use of laboratory animals.