



1. Introduction
Pinctada maxima, commonly referred to as the white butterfly shell, is a Grade II protected species in China and is considered the optimal shell for producing large, high-quality seawater pearls, known as “Nanyang pearls”.1 P. maxima is a tropical and subtropical species with a relatively limited distribution range, primarily found in the Indian Ocean and along the South Pacific coast. In China, it is predominantly distributed in the coastal waters of Hainan Island, the Xisha Islands, and the Leizhou Peninsula.2 It prefers to inhabit groups on coral reefs, shells, rocky substrates, and other marine environments, leading a sessile lifestyle.3 Since the 1980s, wild populations of P. maxima have become progressively scarce and endangered. To facilitate the proliferation of P. maxima, researchers from countries including China, Australia, and Japan have been engaged in artificial breeding efforts, achieving significant advancements.4–6 Although artificial breeding techniques for P. maxima have gradually matured in recent years, the quality of seedlings has declined, leading to significant losses in production operations. The causes of mortality in P. maxima have attracted significant attention from researchers, with numerous studies indicating that some mortality is attributed to limitations in production management, while others are linked to external environmental factors, such as changes in water quality and viral infections. However, the primary cause is the degradation of seedling quality due to gradual inbreeding.7,8 Therefore, to protect and rationally utilize its germplasm resources, it is both necessary and urgent to investigate the genetic diversity of P. maxima.
Currently, research on gene cloning,9 microsatellite marker isolation and screening,10 and genetic diversity analysis of P. maxima11 has emerged as a prominent area of study. Microsatellite markers have become a routine technology in aquaculture, particularly in the study of shellfish germplasm resources and genetic breeding. Microsatellite DNA, also referred to as Short Tandem Repeat (STR) DNA or Simple Sequence Repeat (SSR) DNA, is widely distributed in the genomes of eukaryotes and some prokaryotes.12 Microsatellites exhibit a high degree of variability in the number of repeat units, which may result in variability in the number of repeats or sequence divergence within the repeat units, leading to polymorphism at the locus. Microsatellite DNA is considered one of the most suitable genetic markers for population genetic diversity analysis due to its wide distribution, high polymorphism, reproducibility, conformity to Mendelian inheritance patterns, and co-dominance. Evans et al.13 used microsatellite markers to examine six cultured populations of Haliotis midae from South Africa and Haliotis rubra from Australia, finding a general decrease in genetic diversity across all cultured populations, with the number of alleles reduced by 35-62% compared to wild populations. Han et al.14 used microsatellite markers to analyze the genetic diversity of the Giant Ezo Scallop, employing six microsatellite loci to compare three cultured populations, including the Chinese population and two wild Japanese populations. Du et al.15 employed phenotypic traits, RAPD, and ISSR molecular markers to analyze the genetic diversity of three Perna viridis populations collected from Beihai (Guangxi), Zhanjiang, and Shanwei (Guangdong), China. Although there are many species of economically significant marine shellfish, only a few have been studied using SSR and inter-simple sequence repeat (ISSR) markers, and the application of these molecular techniques in shellfish research remains in its early stages. For P. maxima, Smith et al.16 and Evans et al.13 identified eight and six polymorphic microsatellite DNA markers, respectively. Due to technological limitations at the time, the initial findings on P. maxima require further investigation and refinement. Moreover, most studies focused on artificially bred species, with limited research on the genetic diversity of wild populations. With the rapid advancement of shellfish molecular genetics and biotechnology, the application of SSR markers in population genetic diversity, genetic structure analysis, identification of relatedness and hybrid offspring, genetic map construction, and the evaluation and conservation of P. maxima germplasm resources has become increasingly urgent.
This study utilized a 96-channel fully automated ABI 3730XL genetic analyzer to assess the genetic diversity of three wild P. maxima populations collected from the coastal areas of Danzhou (DZ), Hainan Province, China; Nansha City (NS), Hainan Province; and Xuwen City (XW), Guangdong Province, China. Specific primers were designed based on microsatellite conserved sequences, fluorescent groups were incorporated, fluorescent PCR amplification was conducted, and the amplified products with fluorescent signals were detected via 3730 capillary fluorescence electrophoresis. The fragments with varying numbers of repeat units exhibited peaks at distinct positions on the electropherogram. By interpreting the peak plots, we can identify different alleles, thereby enabling the analysis of the genetic diversity of P. maxima. In conclusion, this study aims to investigate the genetic diversity of three geographic populations of P. maxima using SSR molecular markers, with the objective of revealing their genetic structure, genetic diversity levels, and inter-population differences, as well as providing theoretical insights for the conservation, restoration, and breeding of P. maxima germplasm resources.
2. Materials and methods
2.1. material
Three distinct wild populations of P. maxima were collected from coastal areas at depths ranging from 20 to 30 meters in Danzhou (DZ), Hainan Province; Nansha (NS), Hainan Province; and Xuwen City (XW), Guangdong Province, China. P. maxima were temporarily reared in a land-based recirculating water system at the Sanya Tropical Aquatic Research Institute. The water exchange rate was 100–200% per day, and the water quality parameters were as follows: water temperature ranged from 29 to 30°C, salinity from 32‰ to 33‰, pH between 7.5 and 8.2, dissolved oxygen ≥7.2 mg·L⁻¹, ammonia nitrogen ≤0.2 mg·L⁻¹, and nitrite ≤0.02 mg·L⁻¹.
Fifteen healthy and viable individuals were randomly selected from each of the three P. maxima populations for sampling. Fresh closed-shell muscle tissue of P. maxima was excised and fixed in 70% ethanol for 2 hours, followed by replacement with 95% ethanol, and after 2 to 4 hours, replaced again with 95% ethanol before being stored at -70°C.
2.2. Testing of samples
The samples were analyzed by Wuhan Tianyi Huayu Genetic Technology Co. The analysis process included, in sequence, nucleic acid extraction and detection, typing primer synthesis, fluorescent PCR amplification, primer polymorphism screening, population typing pre-tests, and population typing detection. The experimental reagents were provided by Applied Biosystems, GeneTech, and Axygen.
2.2.1. Synthesis of typing primers
SSR primers were designed based on simplified genome sequence analysis, resulting in the identification of 192 primer pairs for subsequent experiments. The primers were synthesized using the splice method, with a 21-bp splice sequence incorporated into the upstream primer during synthesis. PCR amplification was performed using the junction method, where the upstream primer containing the junction sequence and the downstream primer bind to the template in the first step to generate a PCR product with the junction sequence. In the second step, the junction primer, which contains a fluorescent motif, binds to the downstream primer of the PCR product from the first step to produce a fluorescently labeled PCR product with the 21-bp junction sequence.
2.2.2. Fluorescent PCR amplification
Ten pairs of polymorphic primers were selected to analyze 45 population samples, and PCR reactions were conducted using a Veriti384 PCR instrument (Applied Biosystems). The PCR amplification program was configured as follows: pre-denaturation at 95°C for 5 minutes; denaturation at 95°C for 30 seconds, gradient annealing from 62°C to 52°C for 30 seconds, and extension at 72°C for 30 seconds for 10 cycles; denaturation at 95°C for 30 seconds, annealing at 52°C for 30 seconds, and extension at 72°C for 30 seconds for 25 cycles; followed by an extension at 72°C for 20 minutes, and final storage at 4°C. After completion of the PCR reaction, the amplification products were analyzed using fluorescence capillary electrophoresis (DYY-6C, Beijing Liuyi).
2.3. Data analysis
2.3.1. Analysis of genetic diversity
Various genetic diversity indices for SSR loci and populations, including the number of observed alleles (Na), effective number of alleles (Ne), Shannon’s index (I), polymorphism information content (PIC), observed heterozygosity (Ho), expected heterozygosity (He), and inbreeding coefficient (Fis), were calculated using software such as GenAlEx version 6.501.
2.3.2. Analysis of population genetic structure
Genetic distances between populations were computed using PowerMarker software. Cluster analysis was conducted using the UPGMA method, and a circular dendrogram was generated. Population structure analysis was conducted on 45 samples using STRUCTURE 2.3.4, with K values ranging from 1 to 20, a Burn-in period of 10,000, and 100,000 iterations for the MCMC (Markov Chain Monte Carlo). Each K value was run 30 times, and the optimal ∆K value was calculated using the online tool STRUCTURE HARVESTER. Plots were generated based on the optimal K value obtained from the analysis. The resulting plots from the structural analysis were visualized using CLUMPP and DISTRUCT software.
2.3.3. Analysis of molecular variance (AMOVA) and gene flow estimation
Based on the results of the population genetic structure analysis, the variation and differentiation both between and within populations were calculated and tested for statistical significance using GenAlEx version 6.501 software. The coefficients of genetic differentiation (Fst) and gene flow (Nm) were calculated. Gene flow (Nm) was estimated using Wright’s (1931) formula: Nm = 0.25(1 - Fst) / Fst.
3. Results
3.1. Primer screening results
A total of 192 primer pairs were sourced from the alternative primer library, and 10 primer pairs with a polymorphism information content (PIC) value greater than 0.7 and a minimum allele number of 4 were selected for further analysis. The details of the selected primers are presented in Table 1.
3.2. Genetic polymorphism analysis
The genetic diversity parameters for the 10 microsatellite loci across 45 individuals are summarized in Table 2. A total of 130 alleles (Na) were detected across the 45 samples using 10 primer pairs, with the number of alleles per locus ranging from 8 (minimum) to 17 (maximum), and an average of 13 alleles per locus. The total number of effective alleles (Ne) was 78.798, ranging from 4.874 (PMA036) to 10.33 (PMA164), with an average of 7.8798 effective alleles per locus. The Shannon Index (I) values ranged from 1.774 (PMA036) to 2.513 (PMA164), with a mean value of 2.246. Observed heterozygosity (Ho) ranged from 0.455 (PMA164) to 1 (PMA085), with a mean value of 0.7868. Expected heterozygosity (He) ranged from 0.795 (PMA036) to 0.903 (PMA164), with a mean value of 0.8662. The polymorphic information content (PIC) values ranged from 0.769 (PMA036) to 0.895 (PMA164), with a mean value of 0.8531. According to the criteria outlined by Botstein et al.,17 a PIC value greater than 0.50 indicates that the locus is highly polymorphic. The PIC values of all loci exceeded 0.7, indicating that all loci were highly polymorphic and suitable for effective analysis. The mean inbreeding coefficient was -0.006, with values ranging from -0.199 (PMA085) to -0.410 (PMA164).
The genetic parameters for the three P. maxima populations at 10 microsatellite loci are presented in Table 3. In the NS population, a total of 56 alleles were detected, with the mean number of alleles being 5.6. The effective allele count ranged from 2.1844 to 6.7164, with a mean of 4.0028. Observed heterozygosity ranged from 0.1333 to 1.0, with a mean of 0.6985, while expected heterozygosity ranged from 0.5422 to 0.7959, with a mean of 0.7221. The Shannon Information index ranged from 0.9164 to 2.0375, with a mean of 1.4524. Overall, the NS population exhibited lower mean values for all genetic parameters compared to the other two populations (Table 4).
3.3. Analysis of genetic differentiation
The inbreeding coefficients and gene flow values for the three P. maxima populations at 10 loci are presented in Table 5. The mean inbreeding coefficient (Fis) was -0.006, with a more negative value indicating a low level of inbreeding among the populations. Genetic differentiation between populations is commonly quantified by the genetic differentiation index (Fst) and gene flow (Nm). Only one microsatellite locus, PMA036, had an Fst value ranging from 0 to 0.05, indicating negligible genetic differentiation at this locus in the P. maxima population. The remaining nine microsatellite loci exhibited Fst values ranging from 0.063 to 0.185, indicating significant genetic differentiation at these loci. Since Nm is negatively correlated with Fst, it reflects the reciprocal relationship between population genetic differentiation and gene flow. Among the 10 microsatellite loci, only one locus (PMA036) had an Nm value greater than 4.000, while the remaining eight loci had Nm values between 1.000 and 4.000.
The Fst and Nm values for the three P. maxima populations relative to one another are provided in Table 6. Notably, the Fst between XW and DZ is the highest (7.3878), while the Nm is the lowest (0.033). In contrast, the Fst between NS and DZ is the lowest (2.1400), yet its Nm is the highest (0.105). This suggests a distinct genetic divergence between the XW and NS populations.
According to the results of the analysis of variance (ANOVA) for genetic variation in P. maxima populations (Table 7), The results revealed that the majority of the genetic variation in the three P. maxima populations (90%) originated from individuals, whereas only 10% of the variation was attributed to differences between populations. Thus, individual variation constitutes the primary source of genetic variation in P. maxima.
3.4. Genetic distance and cluster analysis
Principal Coordinate Analysis (PCoA) presents visual coordinates of similarities or differences in research data, and is a non-constrained method of dimensionality reduction analysis of data that can be used to study similarities or dissimilarities in the composition of sample populations.PcoA analysis can reflect differences between 2 groups of samples by visually comparing the straight-line distances between samples in the axes. PCoA was conducted on the three populations, with the results displayed in Figure 1. The NS population is positioned farther from both DZ and XW along the principal coordinate axes, suggesting greater genetic differentiation. In contrast, the closer proximity of DZ and XW along the coordinate axes indicates lower genetic differentiation between the two populations.

The population structure of the 45 samples was analyzed using 10 molecular markers. The ΔK method of structure analysis plots the change in K values as shown in Figure 2. Based on the principle of maximum likelihood, the optimal value of K was determined to be 2 , which resulted in the 45 samples being divided into two distinct subpopulations. Figure 3 represents the STRUCTURE results for 45 samples when K=2. It is clear from the figure that out of the 45 samples, 15 of them contain 14 NS individual samples and 1 XW individual sample grouped into one subspecies, while the remaining 30 samples are grouped into one subspecies.


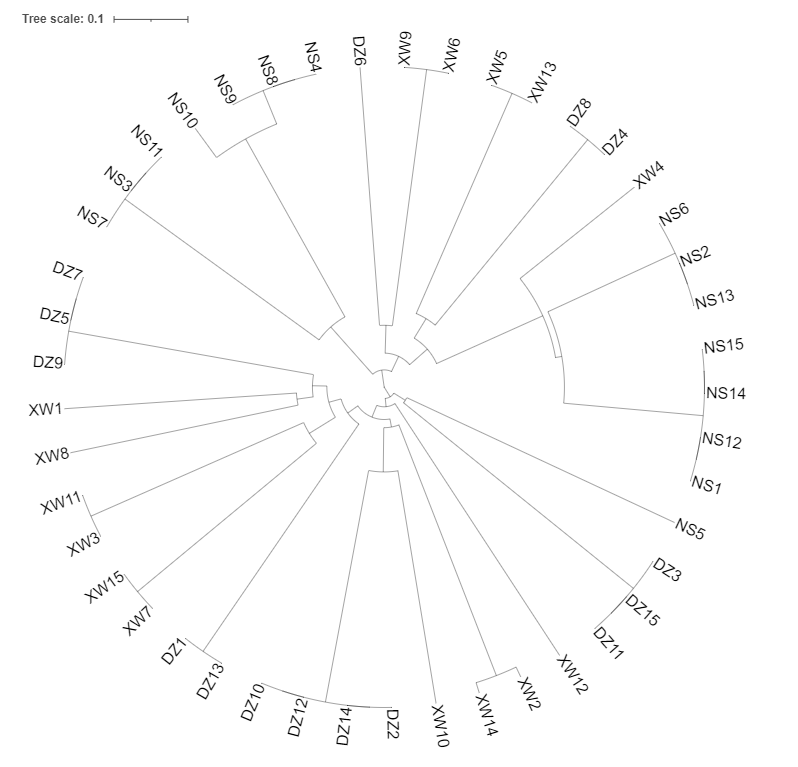
UPGMA clustering analysis was performed based on genetic distances among the P. maxima populations using MEGA 5.0, and the results (Figure 4) revealed that XW and DZ clustered together, while NS formed a distinct group. The UPGMA clustering analysis of the 45 sample individuals is depicted in Figure 5. The analysis reveals that XW and DZ are genetically similar and closely related, while NS is more genetically distant from the other two populations.


4. Discussion
4.1. Transcriptome SSR site screening
The continuous advancement of transcriptome sequencing technology has led to increased sequencing coverage and reduced sequencing costs, making it a crucial tool for the large-scale and high-throughput development of SSR markers. In contrast to traditional SSR marker development methods, transcriptome sequencing technology offers advantages such as reduced time consumption, large data volume, and high efficiency, and has been successfully applied to SSR marker development in aquatic animals. In this study, a 96-channel fully automated ABI 3730XL Genetic Analyzer was used, recognized as the gold standard platform for genetic analysis. This marks the first use of this platform in the genetic analysis of different P. maxima populations. In this study, 192 primer pairs were obtained from the alternative primer library, and 10 SSR loci with a PIC value greater than 0.7 and more than 3 alleles were selected. Compared to Gu et al.,18 who used traditional SSR marker development methods to screen six loci for genetic diversity analysis of P. maxima populations, the polymorphic loci identified from transcriptome data are more accurate and efficient. These loci may be associated with functional genes, providing strong support for subsequent analyses of growth trait associations, genetic map construction, and QTL localization.
4.2. Comparison of genetic diversity within populations of P. maxima
Genetic diversity within a population is typically assessed using metrics such as polymorphic information content (PIC), allele number, and heterozygosity. Polymorphic information content (PIC) serves as a reliable indicator of population diversity. It is widely accepted that a locus is highly polymorphic when PIC > 0.5, moderately polymorphic when 0.25 < PIC < 0.5, and lowly polymorphic when PIC < 0.25.19 In this study, the PIC values of the 10 SSR primers in the three P. maxima populations ranged from 0.769 to 0.895, with a mean value of 0.853, suggesting that the genetic diversity of these populations is high. Regarding the number of alleles, the 10 loci in the three populations (ranging from 8 to 17 alleles) exhibited fewer alleles compared to the Australian wild population (14-68 alleles),16 suggesting a loss of genetic diversity in the Chinese coastal populations of P. maxima. This loss may be linked to the decline in P. maxima resources in China in recent years.
The Hardy-Weinberg equilibrium index (F) characterizes the equilibrium between observed and expected heterozygosity. A positive F-value indicates an excess of heterozygotes, while a negative F-value suggests a deficiency of heterozygotes; the closer F is to 0, the closer the genotype distribution approximates Hardy-Weinberg equilibrium.20 In this study, the F-values of the three populations ranged from -0.0323 to 0.0583, indicating that their genotype distributions are relatively close to Hardy-Weinberg equilibrium. Both the DZ and XW populations exhibited negative F-values, indicating a deficiency of heterozygotes. Previous studies have suggested that heterozygous deficiencies may be associated with null alleles, genotyping errors, sex-linked loci, small sample sizes, and inbreeding.21–23 The two wild populations (DZ and XW) are geographically isolated, suggesting that their heterozygous deficiencies are unlikely a result of inbreeding. The main reason for this phenomenon may be the alteration of the genetic structure of the wild populations of P. maxima due to the massive decrease in the number of wild populations of P. maxima as a result of environmental changes in the sea area and anthropogenic fishing, among others. This occurs in many aquatic animal groups, such as ctenophore scallops,24 turbot,25 carp,26 and Atlantic salmon.27 The mean expected heterozygosity (0.8662) was higher than the observed heterozygosity (0.7868), suggesting that genetic traits are primarily driven by genetic variation and that the population exhibits high genetic diversity. This finding is consistent with the results of Su et al.,28 who analyzed the genetic diversity of P. maxima from Hainan Island, China. Therefore, wild P. maxima from the three regions could be selected to construct target groups with desirable traits, thereby improving seedling quality through continuous optimization.
4.3. Genetic Distance and Genetic Differentiation in of P. maxima
Fst is a crucial parameter that reflects the extent of genetic differentiation among populations. In this study, the mean Fst value for the three P. maxima populations was 0.098, indicating that 9.8% of the genetic differentiation originated from inter-population variation, while 80.2% arose from individual variation, suggesting a moderate level of population differentiation. Hamrick et al.29 argued that when Nm > 1.000, it counteracts the effects of genetic drift and prevents differentiation between populations, whereas when Nm < 1.000, genetic drift can dominate and drive changes in population genetic structure. The results of this study showed that the average Nm among the three P. maxima populations was 2.804, indicating that genetic drift did not dominate changes in the populations’ genetic structure. This suggests that heritable traits across populations tended to homogenize, and the differentiation index among the populations decreased. Furthermore, molecular ANOVA results indicated that 10% of the genetic variation occurred between populations, while 90% was attributed to variation within individuals, with individual variation being the primary source of total genetic variation in the three P. maxima populations. Crawford and Littlepohn30 noted that the timing of population differentiation is a key determinant of variation among populations, and genetic distance serves as an objective indicator of the timing of differentiation and the extent of genetic variation. The maximum genetic distance detected among the three P. maxima populations was 0.6462, and the minimum genetic distance was 0.3352, which are significantly greater than the maximum (0.294) and minimum (0.026) genetic distances reported by Su et al.28 Modern hybrid dominance theory suggests that hybrid vigor in offspring is positively correlated with genetic variation among parents.31 Therefore, in the artificial breeding of P. maxima, individuals with desirable traits from genetically distant populations should be selected as parents to improve seed quality and expand the parental gene pool.
4.4. Molecularly assisted population breeding of P. maxima
Population selection technique is a phenotypic consistency selection to make the genotypes tend to be the same is to increase the degree of purity, to establish varieties with excellent traits, and then through the establishment, maintenance of different sources of populations and often in two or more populations between the exchange of parentage to control inbreeding, to maintain a certain level of heterozygosity of varieties or strains.32 Understanding the genetic background between different populations plays a very important role in seed conservation and establishing further selection programmes. Studies have shown that the use of microsatellite markers developed to assess the genetic diversity of parental populations and to evaluate genetic differences between individuals at the molecular level can be effective in guiding the establishment of family lines and avoiding inbreeding.33,34 In this study, the UPGMA clustering results of the three populations revealed that the XW and DZ populations had the smallest genetic distances from each other and clustered into one group, while the NS population clustered into a separate group.In terms of geographic distribution, the XW and DZ populations are located at similar latitudes, although they come from different seas, and their growth traits and genetic backgrounds are more similar because the two populations are subjected to similar external environmental factors, such as climate and temperature, for their growth and reproduction. Gale et al.35 found that in localised waters at the same latitude, especially around islands or closed bays, gene flow between populations is limited due to current constraints, resulting in reduced genetic diversity. This also confirms the viewpoint of this paper. The NS population is geographically isolated, and its growth environment is in the tropical sea, so its genetic diversity is relatively high.This is in line with the findings of this study. Hedgecock et al.36 found that gene flow may be more frequent in warmer tropical regions where water temperatures are higher and biodiversity is abundant, resulting in higher genetic diversity. This is consistent with the results of this study. Combined with the fact that the He of the XW population was higher than that of the DZ population, this indicates that the XW and NS population has relatively greater potential for variety selection. However, in order to further enrich the genetic diversity of the populations, improve and enhance the viability and adaptability of the populations, and avoid the inbreeding decline of the populations, we can prioritise the NS and XW populations as the base breeding groups.
Declaration
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
The study was supported by Key Research and Development Plan Projects in Hainan (ZDYF2021XDNY132,Distant Sea Aquaculture Technology and Species Development Innovation Team,the earmarked fund for CARS (CARS-49,South China Sea Staple Economic Species Disease and Ecological Prevention and Control Innovation Team, Sanya Agricultural Science and Technology Innovation Project (2019NK13).
Author Contributions
Formal Analysis: Wei Fang (Equal), Wang Zhao (Equal), Yu Wang (Equal). Writing – original draft: Wei Fang (Lead). Methodology: Mingqiang Chen (Lead). Writing – review & editing: Mingqiang Chen (Lead). Investigation: Wang Zhao (Equal), Yu Wang (Equal). Conceptualization: Zhenhua Ma (Lead). Supervision: Zhenhua Ma (Lead).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.