

Introduction
E. fuscoguttatus♀ × E. lanceolatus♂ is an important marine Epinephelus species, it has a faster growth rate, greater disease resistance, and better tolerance to captive conditions than its parent varieties.1 Given these biological advantages, E. fuscoguttatus♀ × E. lanceolatus♂ would be ideally suited for large scale breeding in coastal areas of Asia.2 It has become one of the most important mariculture fish in China and Southeast Asian countries. However, with the continuous expansion of farming and production scale, intensive Epinephelus farming is plagued by infectious diseases caused by viruses, bacteria, fungi and parasitic pathogens.3 Probiotics are regarded as environment-friendly feed supplements and have been confirmed to have good effects in disease control.4
Water is a key factor for the probiotic in aquaculture systems. The health of fish cultured in aquaculture systems mainly depends on water quality, including oxygen levels and contamination with chemical or biological pollutants.5 Microorganisms are important colonizers and play vital roles in the sediments and water of various natural environments.6 Many bacterial species are involved in the degradation of organic matter and have been found to participate in nitrogen,7,8 phosphorus cycles,9 as well as other biogeochemical processes. Moreover, eukaryotic microorganisms, including protists, algae, unicellular fungi, and small invertebrates, are involved in a variety of ecological functions in water and sediments, including biogeochemical cycles and food network interactions.10,11 Different microbial taxa appear to have specific preferences for sediment and water environments. Dai et al.12 compared eukaryotic and bacterial microorganisms in sediment and water across aquaculture ponds in different regions of China. The analysis revealed significantly higher microbial abundance in aquatic environments, particularly within the Cyanobacteria and Actinobacteria phyla, Betaproteobacteria and Alphaproteobacteria classes, Roseiflexaceae and Cryomorphaceae families, and Dinghuibacter genus.12 Long-term microbiological water monitoring in the Volga Delta showed that bacteria of the family Enterobacteriaceae and genera Aeromonas, Pseudomonas, and Flavobacterium dominated the microflora of water with a rich species composition. Mycobiota is represented by fungi of the genera Aspergillus, Fusarium, and Candida.13
Recirculating aquaculture systems are commonly used in fish farming and rely on microorganisms to maintain proper water quality, nutrient cycling, animal welfare, and disease control. However, feeding operations in fish farms may negatively affect the community composition of microorganisms and create a favorable environment for opportunistic pathogens.14 Currently, the understanding of microbial communities within recirculating aquaculture systems of Epinephelus farming is scarce, and no relevant studies have been reported in Hainan, which presents an obstacle to proactive system management. Particularly, microbiota in different kinds of recirculating aquaculture systems has great significance for the use of probiotics in these environments. In this study, aquaculture water samples were collected from marine circulation aquaculture farms in Wenchang, Wanning, Lingshui, Ledong, and Dongfang along the coast of Hainan, and bacterial and fungal flora in the cultured water were determined to elucidate the spatial and temporal structure of microbial communities in Epinephelus farms. The distinct characteristics of microbial communities in the cultured water of E. fuscoguttatus♀ × E. lanceolatus♂ were analyzed by comparing the species of bacteria and fungi in different aquaculture systems for the guidance of probiotics.
Materials and Methods
STUDY SITE AND SAMPLE COLLECTION
Hainan Island is located in Southern China. Huiwen, Gangbei, Lian, Huangliu and Gancheng evenly distributed along the coast of Hainan. Water samples were collected from E. fuscoguttatus♀ × E. lanceolatus♂ seawater recirculation systems at these five locations. The mariculture ponds in Huiwen and Gangbei are outdoors, while those in Lian, Huangliu and Gancheng are indoor marine fish culture systems. The samples were labeled with acronyms corresponding to their collection sites: FWHW (Huiwen), FWGB (Gangbei), FWLA (Lian), FWHL (Huangliu), and FWGC (Gancheng). Water samples were systematically collected from three distinct aquatic systems within the same geographical region. At each system, sampling was performed 0.5 meters from the pond edge at a consistent depth of 0.3 meters using pre-sterilized beakers. Each sample was obtained by combining water collected from five sampling points that were thoroughly mixed in bottles. Following collection, samples were immediately sealed in sterile containers, stored in ice boxes, and transported to the analytical laboratory. Water samples were filtered with 0.45 μm sterile filter membranes, and the microorganisms-enriched filter membranes were packed into sterilized centrifuge tubes and stored at -80℃ for DNA extraction.
DNA EXTRACTION, PCR AMPLIFICATION AND SEQUENCING
Total genomic DNA was extracted from the filter membranes using the OMEGA Soil DNA Kit (M5635-02; Omega Bio-Tek, Norcross, GA, USA) in accordance with the manufacturer’s protocol. Extracted DNA were measured using a NanoDrop NC2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and agarose gel electrophoresis, respectively.15 Primer 338F (5’-ACTCCTACGGGAGGCAGCA-3’) and primer 806R (5’-GGACTACHVGGGTWTCTAAT-3’) were used to amplify the 16S rDNA V3–V4 region.16 Primers ITS5 (5’-GGAAGTAAAAGTCGTAACAAGG-3’) and primer ITS2 (5’-GCTGCGTTCTTCATCGATGC-3’) were used to amplify the ITS1 region.17 To enable multiplex sequencing, sample-specific 7-bp barcodes were incorporated into the forward and reverse primers. PCR mixture was prepared in a total volume of 25 μL containing the following components: 5 μL of 5 × reaction buffer, 0.25 μL Fast pfu DNA Polymerase (5 U/μL), 2 μL dNTP mixture, 1 μL each of forward and reverse primers (10 μM), 1 μL DNA template, 14.75 μL sterile ddH₂O. PCR amplification and product purification were performed as previously described by Chen et al.18 Illumina NovaSeq platform with NovaSeq 6000 SP Reagent Kit (500 cycles) at Shanghai Personal Biotechnology Co., Ltd (Shanghai, China) was used for sequencing.
SEQUENCES, BIOINFORMATICS AND STATISTICAL ANALYSIS
Sequencing data were processed using QIIME 2 with slight modifications to the standard method.19 DADA2 plugin was used for sequence primer removal, quality filtering, denoising, merging, and chimaera removal.20 Non-singleton amplicon sequence variants (ASVs) were aligned using MAFFT.21 The classifier-sklearn naïve Bayes classifier in the feature-classifier plugin22 was applied to classify ASVs for bacteria and fungi against the Greengenes/UNITE Release 8.0 database, respectively.23,24
Sequence data were analyzed using the QIIME2 and R packages (v3.2.0). Chao1, Shannon, Simpson, Faith’s PD, Pielou’s evenness, and Good’s coverage were calculated using the ASV table in QIIME2, and they were visualized as box plots. Beta diversity analysis was performed to investigate the structural variation of microbial communities across samples using Bray-Curtis metrics.25 Alpha and beta diversity were visualized via box diagrams and principal coordinate analysis (PCoA), respectively. Significant differentiation of microbiota structure among groups were assessed using QIIME2 based on analysis of similarity (ANOSIM).26,27 Venn diagram was constructed using the R package “Venn Diagram” to visualize the shared and unique ASVs across samples. The analysis was based on ASV presence data rather than relative abundance values.28 Taxa abundance at the phyla and genus levels was statistically compared among samples using Origin 2024. The default parameters were used to detect differentially abundant taxa across groups based on the Linear discriminant analysis effect size.29 Co-occurrence analysis was performed using SparCC analysis. Pseudocount value in SparCC was set to 10-6. The cutoff of correlation coefficients was determined as 70 through random matrix theory-based methods as implemented in R package RMThreshold. Co-occurrence network with nodes representing ASVs and edges representing correlations between these ASVs was constructed based on the correlation coefficients. iGraph was then used to construct the correlation network diagram.
Results
MICROBIAL COMMUNITY DIVERSITY
The rarefaction curves of all five samples were relatively stable and smooth, indicating that the number of ASVs adequately represented the bacterial and fungal communities. The sequencing depth reflects the structural characteristics of the microorganisms collected at the five different local sites. Good’s coverage value of bacteria in water samples was above 0.970, and that of fungi was above 0.999, indicating that the sequencing results properly represented the real situation of microorganisms in water samples at different sampling points (Fig. 1 A). Among the five samples, Chao1, Faith_pd, Shannon, Pielou’s, and Observed species indices of bacteria showed significant differences (Kruskal-Wallis test, P < 0.05). In contrast, fungi showed no significant differences in other indices except Pielou’s index (Fig. 1 B). The results showed that the diversity of bacteria in the five samples was relatively high, whereas that of fungi was not.
_and_fungal_(b)_-diversity_indices_of_the_water_from_fwhw__fwgb__fwla__f.png)
MICROBIAL COMMUNITY DIFFERENTIATION
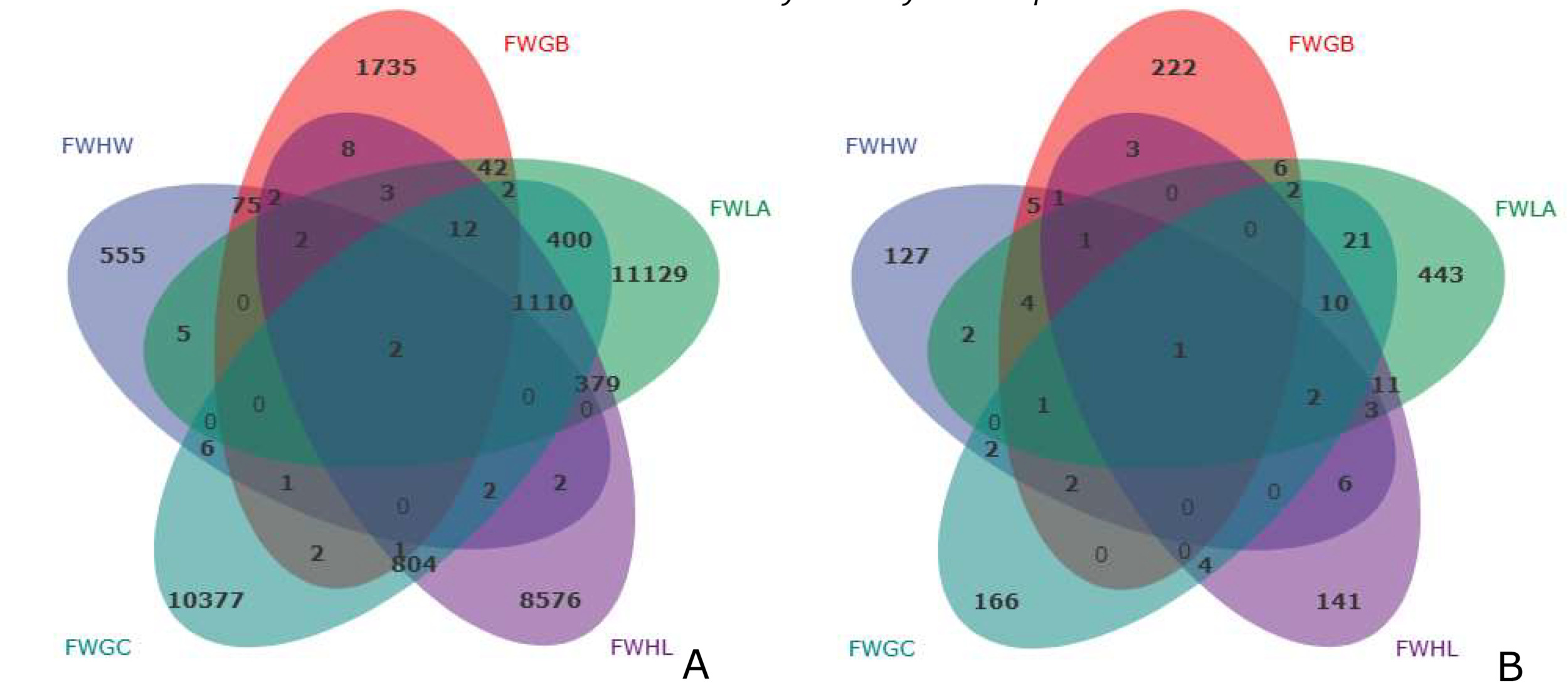
Venn diagrams showed 555, 1,735, 11,129, 8,576, and 10,377 unique bacterial ASVs in the FWHW, FWGB, FWLA, FWHL, and FWGC samples, respectively. In contrast, there were 127, 222, 443, 141, and 166 unique fungal ASVs in these five samples, respectively (Fig. 2). In addition, two bacterial ASVs and one fungal ASV were separately identified in all samples at the five sites. As for why the five locations shared few common bacterial and fungal ASVs, we will speculate on the possible effects of geography in our subsequent reports.
_and_fungal_(b)_asvs_distribution_in_aquaculture_water_fr.png)
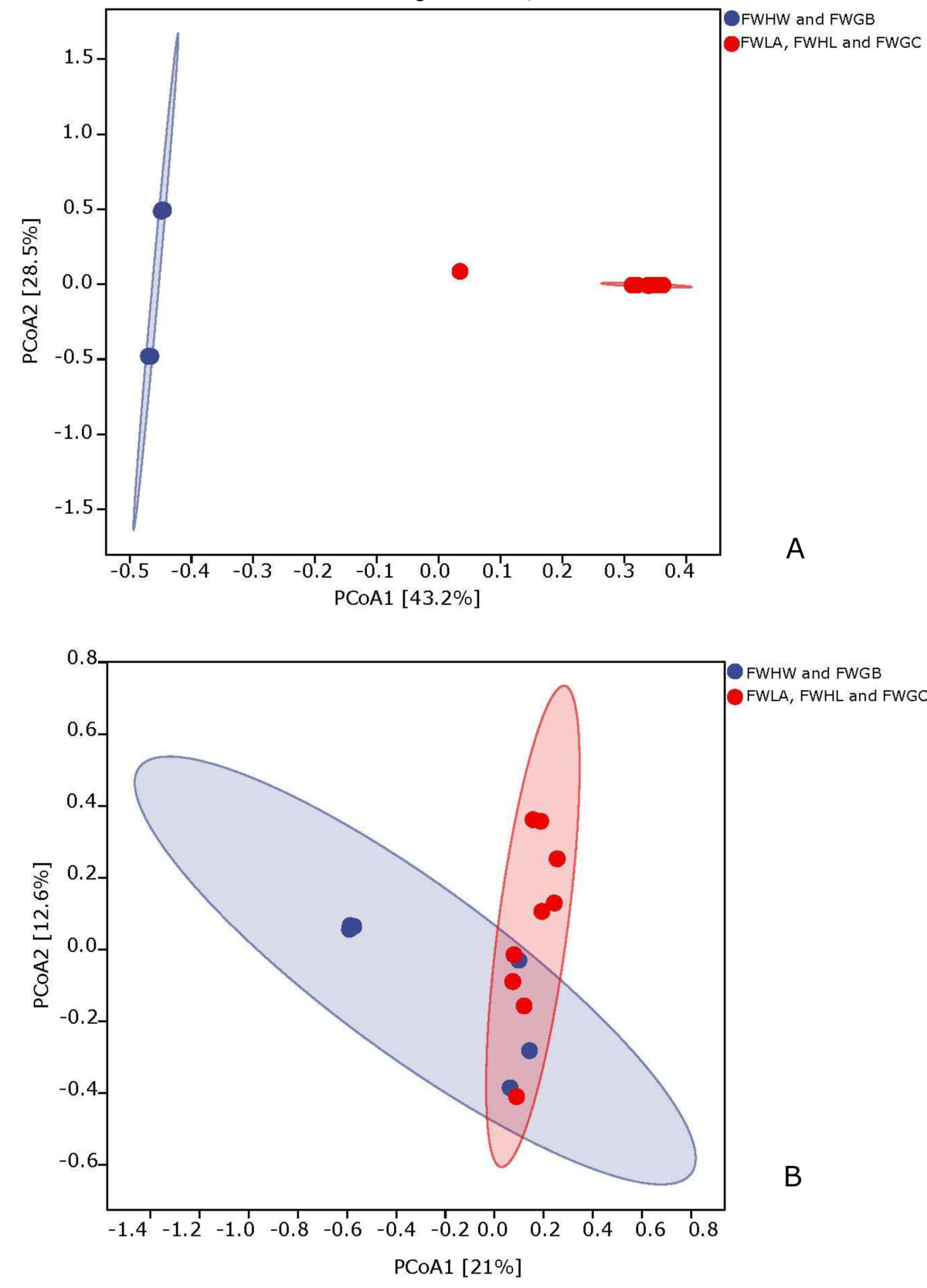
According to the PCoA analysis based on the Bray-Curtis distance, the first two principal components of the bacteria accounted for 43.2% and 28.5%, respectively (Fig. 3A). The fungi accounted for 21% and 12.6%, respectively (Fig. 3B). According to the ANOSIM analysis, there were significant differences in bacterial and fungal structure between FWHW, FWGB vs FWLA, FWHL, and FWGC samples (P < 0.05). These results indicated that the microorganisms existing in water are significantly different in the outdoor mariculture ponds and the indoor marine fish culture system.

MICROBIAL COMMUNITY COMPOSITION AND STRUCTURE
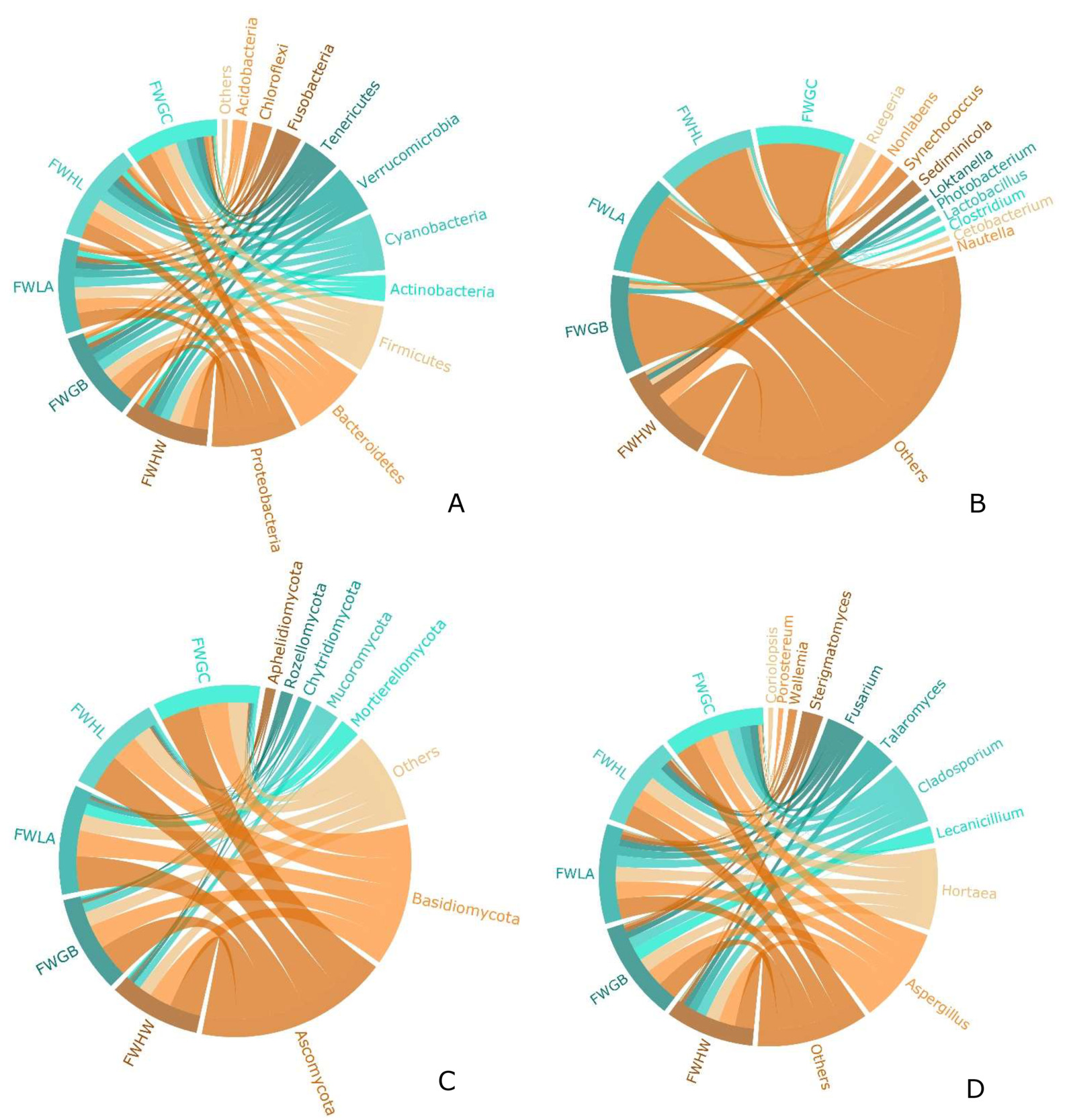
Figs. 4A and 4B show the composition of the bacterial communities at the phylum and genus levels. The dominant bacteria at the five sites were Proteobacteria, Bacteroidetes, Firmicutes, Actinobacteria, and Cyanobacteria. Compared with the other four samples, among the top 10 dominant bacteria in the FWHW sample, Proteobacteria and Bacteroidetes had the highest relative abundances. In contrast, Firmicutes and Actinobacteria had the lowest relative abundances. The relative abundance of Actinobacteria peaked in the FWGB sample, whereas the proportion of Cyanobacteria in the FWLA sample was higher than that in other samples. Ruegeria and Loktanella from Proteobacteria were present in all five samples. In addition to the FWHW sample, Synechococcus from Cyanobacteria was present in the other four samples. It showed that Lactobacillus in Acidobacteria were present in a certain percentage of the FWLA, FWHL, and FWGC samples.
Figs. 4C and 4D show the composition of the fungal communities at the phylum and genus levels. Ascomycota and Basidiomycota were predominant in all the samples. In the comparison of the top seven dominant fungi, the proportion of Ascomycota in the FWHW and FWHL samples was the highest, accounting for 0.8922±0.0432% and 0.8949±0.0243% of the fungal communities, respectively. However, the proportion of Basidiomycota in the FWHW and FWHL samples was the lowest, accounting for 0.0718±0.0743% and 0.0865±0.0255% of the fungal communities, respectively. Furthermore, Aspergillus and Hortaea accounted for the maximum proportion in the FWHL sample, while Lecanicillium was only present in FWGB sample, accounting for 0.5236±0.0742% of the fungal community.
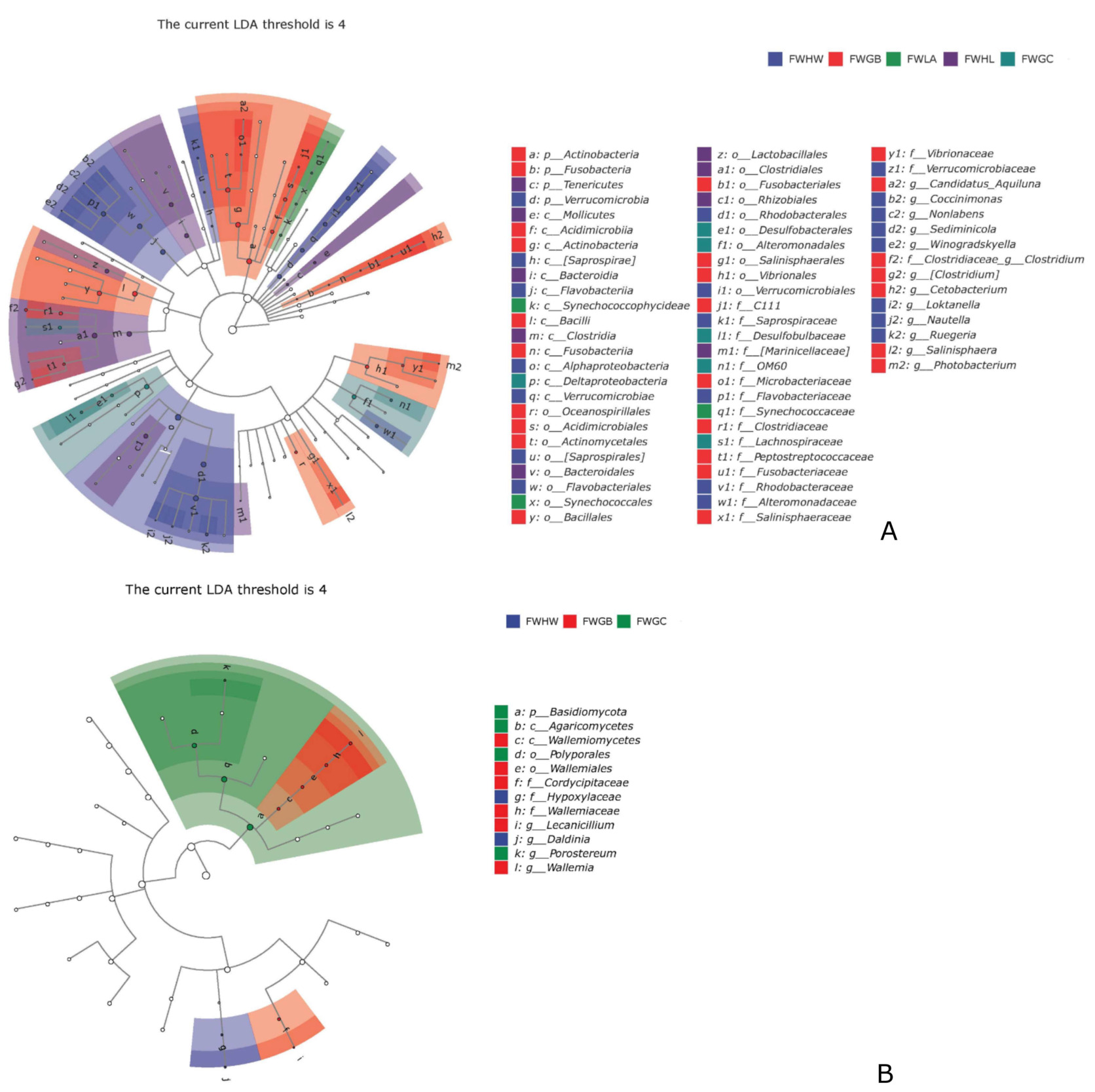
LEfSe was used to analyze further bacterial and fungal genera in the water of the outdoor mariculture ponds and the indoor marine fish culture system. A threshold of 4 was employed for the logarithmic LDA score to determine the discriminative feature. Actinobacteria, Fusobacteria, Verrucomicrobia were hyper-dominant in the water of the outdoor mariculture ponds. Tenericutes were the most dominant phyla in the indoor marine fish culture system (Fig. 5A). The bacterial genera Candidatus_Aquiluna, Coccinimonas, Nonlabens, Sediminicola, Winogradskyella, Clostridiaceae_Clostridium, [Clostridium], Cetobacterium, Loktanella, Nautella, Ruegeria, Salinisphaera and Photobacterium were more abundant in the outdoor mariculture ponds. As for fungi, Basidiomycota were more abundant in the water of sample collected from the indoor marine fish culture system of Gancheng. Simply, the genera Lecanicillium, Daldinia and Wallemia were dominant in the water of outdoor mariculture ponds, whereas Porostereum was the only genus typical dominant in the indoor marine fish culture system (Fig. 5B).

_and_fun.png)
RELATIONSHIP BETWEEN ABUNDANCE OF CLOSTRIDIUM AND BACTERIAL INDICATORS
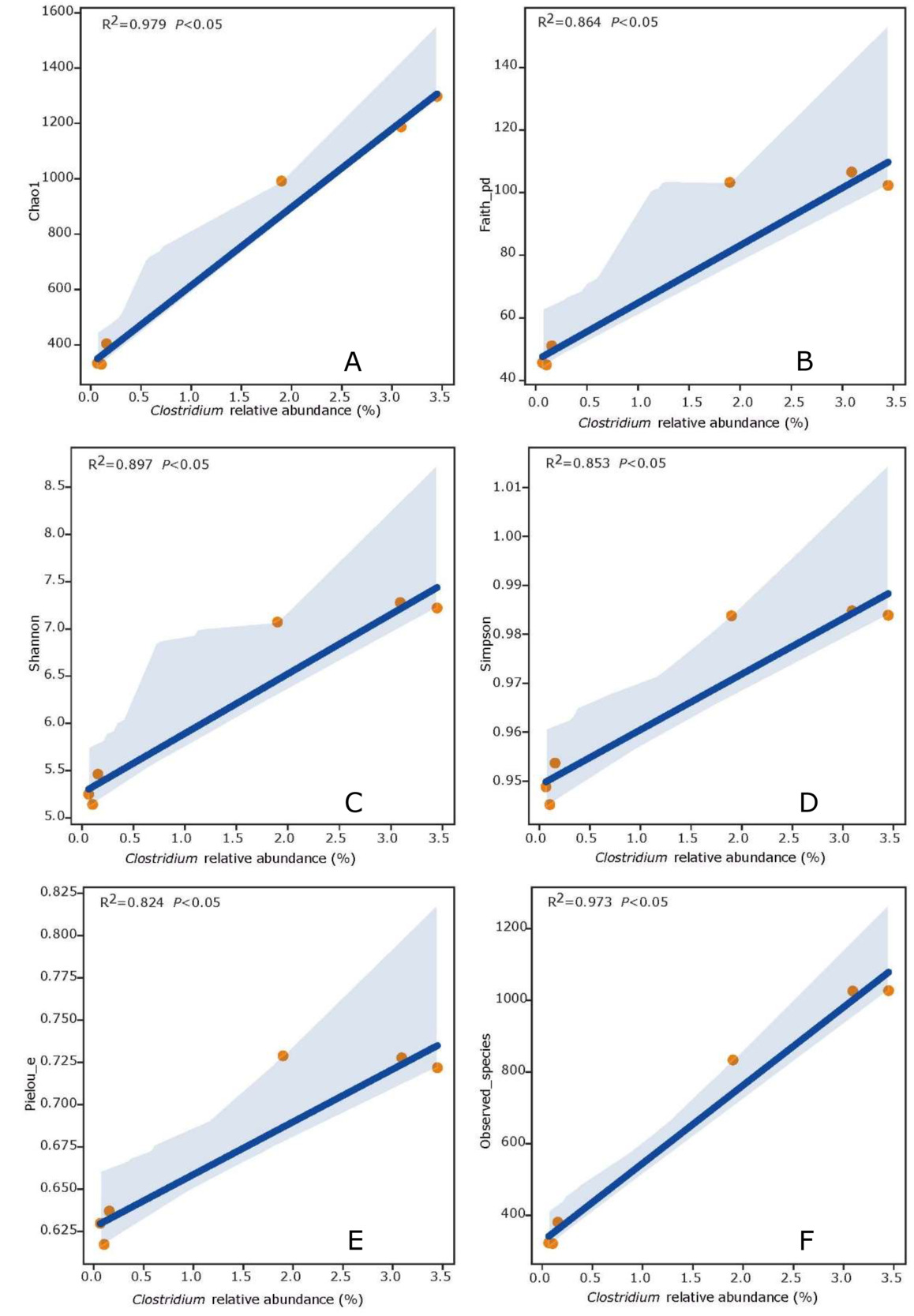
The Chao1, Faith_pd, Shannon, Simpson, Pielou_e and Observed species of the outdoor mariculture ponds were selected as bacterial indicators in the linear model, as they could explain the relationship with Clostridium abundance. As a result, Chao1 (R2 = 0.979, P < 0.05), Faith_pd (R2 = 0.864, P < 0.05), Shannon (R2 = 0.897, P < 0.05), Simpson (R2 = 0.853, P < 0.05), Pielou_e (R2 = 0.824, P < 0.05) and Observed species (R2 = 0.973, P < 0.05) had a significant positive relationship with Clostridium abundance. (Fig. 6).
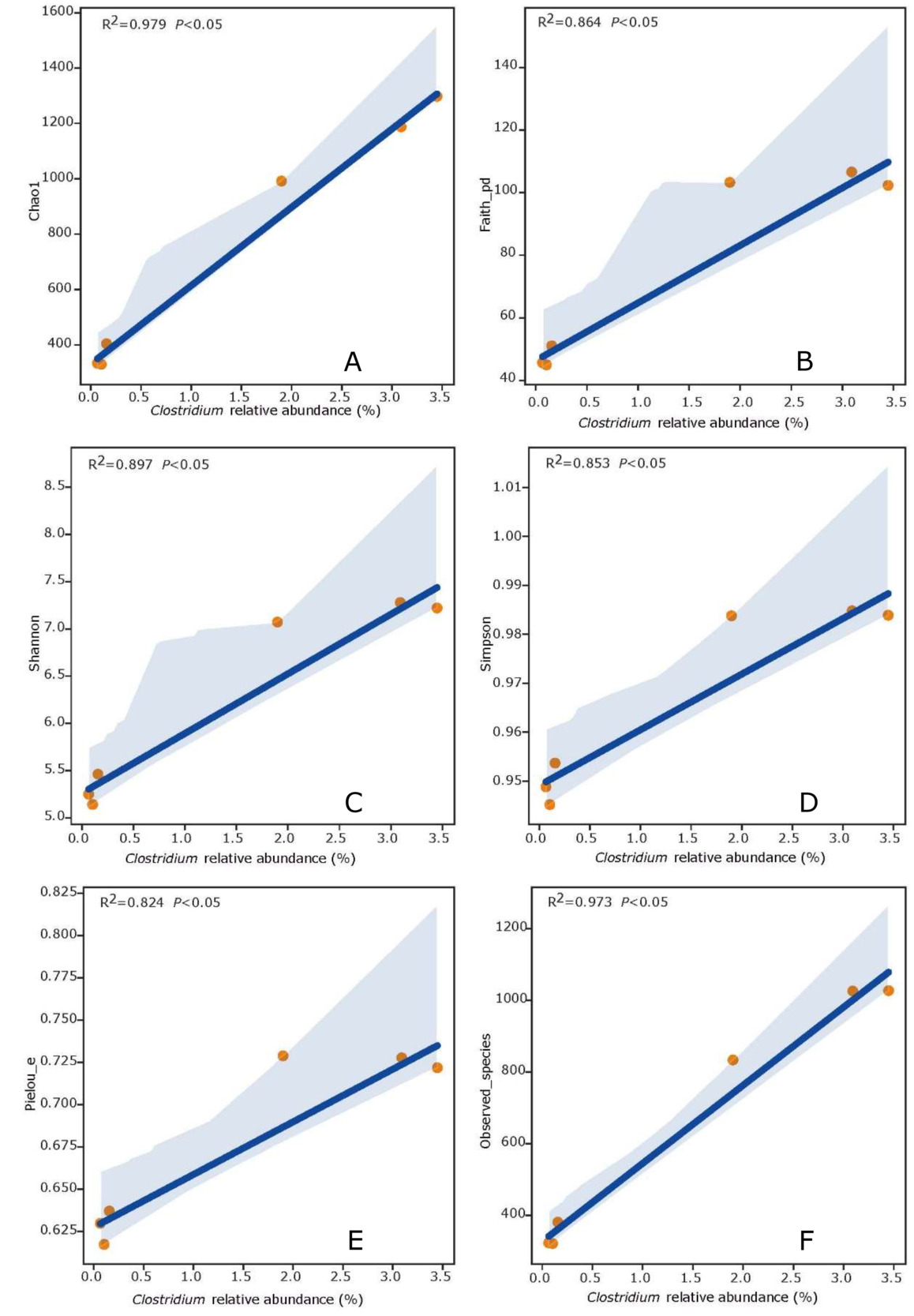
MICROBIAL COMMUNITY NETWORKS
Microbial interactions in the five samples of FWHW, FWGB, FWLA, FWHL and FWGC were investigated using a general-level network analysis (Fig. 7). Connections between nodes indicate that the genera are related, and the higher the index of edge through a node, the higher the association of the genus with other genera. We selected the co-occurrence and co-exclusion genera related with Clostridium from the top 45 abundance bacterial genera. Clostridium was positively correlated with Synechococcus, Formosa, Antarctobacter and [Clostridium]. However, it was negatively correlated with Nautella, Ruegeria, Haloferula, Roseovarius and Loktanella. It provided a possible way to regulate the abundance of Clostridium through microbial interaction in the future.
Discussion
Studies on microbial communities in aquatic environments where fish live provide reliable clues regarding the hygiene status of the environment because water quality is closely linked to fish diseases.30 Detecting fish pathogenic bacteria, including novel bacteria, and changes in water microbiota are important indicators of environmental pollution. In this study, the bacterial species diversity was significant different in cultured water samples of E. fuscoguttatus♀ × E. lanceolatus♂ from the Wenchang, Wanning, Lingshui, Ledong and Dongfang coastal areas, whereas that of fungi was not significant. Analysis of the classified taxa revealed that bacterial communities in aquaculture water were mainly dominated by Proteobacteria, Bacteroidetes, Firmicutes, Actinobacteria, and Cyanobacteria, many of which have been reported to be highly active in the decomposition of organic matter.31,32 However, Ascomycota and Basidiomycota were the main fungi in the water samples but were not involved in organic decomposition and photosynthesis. Considering the abundance and diversity of bacteria, bacterial communities in the water column may be important for organic decomposition and photosynthesis. In addition, the species structure of bacteria in the five samples was relatively different, whereas that of fungi was not significantly different. Although different taxa were detected in the aquaculture water, the fungal richness and diversity were not significantly different, and the fungal communities in the water samples shared some similarities. Environmental filtering and dispersal limitation were considered the important factors in shaping the patterns of fungal diversity.33 Fungal taxa in aquatic environments may be less influenced by the heterogeneity differences between water samples than bacteria, considering the physical connection between these five habitats. Specifically, bacteria and fungi have been found to consist primarily of environmental selection and drift, respectively.34 As a result, fungal communities might be more evenly distributed in the aquaculture water.
The water temperature, pH, dissolved oxygen, and nutrient levels all showed significant influence on the microbial communities in fish farming water.12 The intestinal microbial community structure of Penaeus japonicus was similar to that of water in culture ponds.35 Zeng et al.36 demonstrated that Hypophthalmichthys molitrix, Aristichthys nobilis, Ctenopharyngodon idellus and Carassius auratus have different prevalent microbiota. There is a strong correlation between fish intestinal microbiota and the water environment, and the fish with a weak correlation is Hypophthalmichthys molitrix. These studies showed the microbial community characteristics of farming water and indicated a strong relation with intestinal health. Here, we found that Clostridium was more abundant in the outdoor mariculture ponds, and it was the marker strain in the bacterial community. Previous studies have confirmed that diets supplemented with C. butyricum could enhance digestive and absorption ability in aquatic animals and could also prevent or diminish the intestinal inflammatory response. Furthermore, it could enhance intestinal epithelial barriers and regulate the structure of intestinal microflora, and it would inhibit pathogenic bacteria.37 It indicated Clostridium of aquaculture water was important for the intestinal health of E. fuscoguttatus♀ × E. lanceolatus♂. Interestingly, we found that Clostridium was positively correlated with Synechococcus. It provided a potential way to regulate the abundance of Clostridium. We could use Synechococcus to synergistically and positively regulate the abundance of Clostridium.
The intestinal amylase and protease activities and the expression of digestive genes increased in Litopenaeus vannamei when fed with C. butyricum.38,39 The growth, immunity and intestinal microbiota improved in Cherax cainii when fed with C. butyricum.40 Similarly, the tilapia research indicated that C. butyricum enhanced growth, improved immune response, regulated intestinal microbiota and elevated disease resistance.41 These studies revealed that an appropriate supplement of C. butyricum in diets could enhance digestive functions, prevent the intestinal inflammatory response, and regulate the structure of intestinal microbiota. We found that the bacterial indicators of the outdoor mariculture ponds showed positive linear relationships with Clostridium abundance. It indicated that the abundance of Clostridium could influence microbial community diversity. Therefore, a probiotic of Clostridium possibly affected the microbial community diversity of aquaculture water when it was added to the outdoor mariculture ponds. Previous studies revealed that moderate C. butyricum improved intestinal morphology and the balance of the intestinal microbiota, as well as increasing beneficial bacteria and decreasing the incursion of opportunistic pathogens to the host. From this study, we could propose that Clostridium was added to the outdoor mariculture ponds as a probiotic; the dosage needs to be controlled appropriately to avoid the excessive change of microbial diversity of water microbiota.
In conclusion, this study analyzed the bacteria and fungi in marine circulation aquaculture waters of E. fuscoguttatus♀ × E. lanceolatus♂ in Wenchang, Wanning, Lingshui, Ledong, and Dongfang to understand the microbiota in the coastal fish aquaculture waters of Hainan. The results showed significant differences in bacterial abundance, species diversity, and community structure in the cultured water. At the same time, there were minimal differentiation in fungi, revealing a possible interspecific relationship between microbiological communities. In addition, the growth of Clostridium in the aquaculture water was positively correlated with the bacterial indicators. Therefore, it is needed to modulate the use dosage of Clostridium to improve the growth performance of E. fuscoguttatus♀ × E. lanceolatus♂ in the outdoor mariculture ponds, avoiding the excessive change of microbial community diversity.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32160866).
Authors’ Contribution
Methodology: Minjing Zheng (Equal), Zhen Li (Equal). Writing – review & editing: Minjing Zheng (Equal), Yonggan Chen (Equal). Software: Minjing Zheng (Equal), Yonggan Chen (Equal). Conceptualization: Yonggan Chen (Lead). Project administration: Yonggan Chen (Lead). Funding acquisition: Yonggan Chen (Lead).
Competing of Interest – COPE
The authors declare no conflicts of interest.
Ethical Conduct Approval – IACUC
No animal sampling was performed in this research.
Informed Consent Statement
All authors and institutions have confirmed this manuscript for publication.
Data Availability Statement
Microbial sequence data were deposited in the China National Microbiology Data Center (NMDC) with accession numbers NMDC10018592 and NMDC10018593, respectively.