



Introduction
Mitochondria, ubiquitous in nearly all eukaryotic organisms, serve pivotal roles in regulating energy metabolism, apoptosis, aging, and various diseases, rendering them indispensable cellular components.1 Mitochondrial DNA (mtDNA) emerges as a valuable molecular marker for systematic inquiries, owing to its straightforward structure, accelerated evolutionary pace, copious copies, and straightforward isolation techniques. These attributes render mtDNA a convenient and efficient tool for scrutinizing genetic relationships and delineating phylogenetic frameworks.2 Mitochondrial genomes, essential in molecular biology research, furnish critical insights into evolutionary connections, population histories, and genetic variations.3 Widely utilized in species delineation, classification, and phylogenetic analyses, they unveil species phylogenetic interrelations and aid in constructing evolutionary lineages within a genus.4 Furthermore, mitochondrial genomes facilitate investigations into gene flow, migration routes, and genetic diversity among species.5
Schizothorax, also known as snowtrout or Schizothoracins, belongs to the genus of ray-finned fish in the Cyprinidae family.6 These fish predominantly inhabit the freshwater systems of Asia, especially in high-altitude areas like the Himalayas, Tibetan Plateau, and nearby regions.7 Adapted to cold and high-altitude conditions, species of Schizothorax play a crucial role in maintaining the ecological balance of their habitats. Among the species, Schizothorax biddulphi is particularly noteworthy.8 It flourishes in the mountainous river systems of Asia, characterized by a spindle-shaped body, small mouth, and unique teeth.
However, S. biddulphi is at risk due to increasing human activities,9 habitat degradation,10 the effects of climate change, and emerging pathogen like Acinetobacter lwoffii11 marking it as a species needing urgent conservation efforts.12 Current research focuses on its ecology, conservation biology, and genetics to better understand its habitat requirements, reproductive behaviors, and population dynamics.8,13–15 These studies are essential for developing effective conservation strategies and managing its genetic diversity for future preservation.
This study presents a comprehensive analysis of the mitogenome of S. biddulphi. We have successfully assembled the complete mitogenome of S. biddulphi using Hifi sequencing technology. This research significantly contributes to the phylogenetic understanding of Schizothorax, providing novel insights into the genus and facilitating taxonomic revisions, especially regarding species relationships within this diverse genus. This achievement not only enhances our understanding of S. biddulphi’s genetic composition but also provides valuable insights into the phylogenetic relationships within the broader family Cyprinidae.
Materials and Methods
Sample and Sequence
The fish species featured in this study were collected from Weigan River in Tarim River basin ( 41°38 ‘25 "N, 81°26’ 42" E) of Xinjiang. China, and are housed at Tarim University (Collection No. 20230816006; for inquiries, contact Shengao Chen, shengao@taru.edu.cn) (Figure 1).

Genomic DNA extraction was performed using the TIANamp Genomic DNA Kit (TIANGEN, Beijing, China). The HiFi Library was prepared following the manufacturer’s protocol. Initially, a 15 μg sample was selected and the SMRTbell® Express Template Preparation Kit v2 was employed to construct the SMRTbell library. Subsequently, small DNA fragments were eliminated using BluePippin. The SMRTbell template was then annealed with a sequence primer, and the resulting complex was bound by DNA polymerase. The library underwent sequencing on the Sequel II sequencing platform (Pacific Biosciences of California, Inc., Menlo Park, CA, USA). CCS (v.6.4.0) software was utilized to generate the HiFi reads.
Mitogenome Assembly and Annotation
We constructed the mitochondrial genome of S. biddulphi by employing the MitoHiFi pipeline (https://github.com/marcelauliano/MitoHiFi), which utilized PacBio HiFi reads and was guided by a reference sequence. The initial reference sequence used was from Schizothorax lissolabiatus (NCBI: NC_027162.1). Following the assembly of the nuclear genome, we employed BLAST+ (Camacho et al. 2009) to identify similarities between the mitochondrial assembly and the nuclear genome. To exclude nuclear DNA contamination (NUMTs), we compared the mitochondrial assembly against the nuclear genome using BLAST+ with a 99% sequence identity threshold. Nuclear contigs and scaffolds with more than 99% identity to the nuclear genome were discarded. To quantify sequencing depth and validate the assembly, HiFi reads were mapped back to the final circular mitogenome using minimap2 with the HiFi preset (-ax map-hifi), and alignments were sorted and indexed with samtools. Per-base depth was computed with samtools depth.
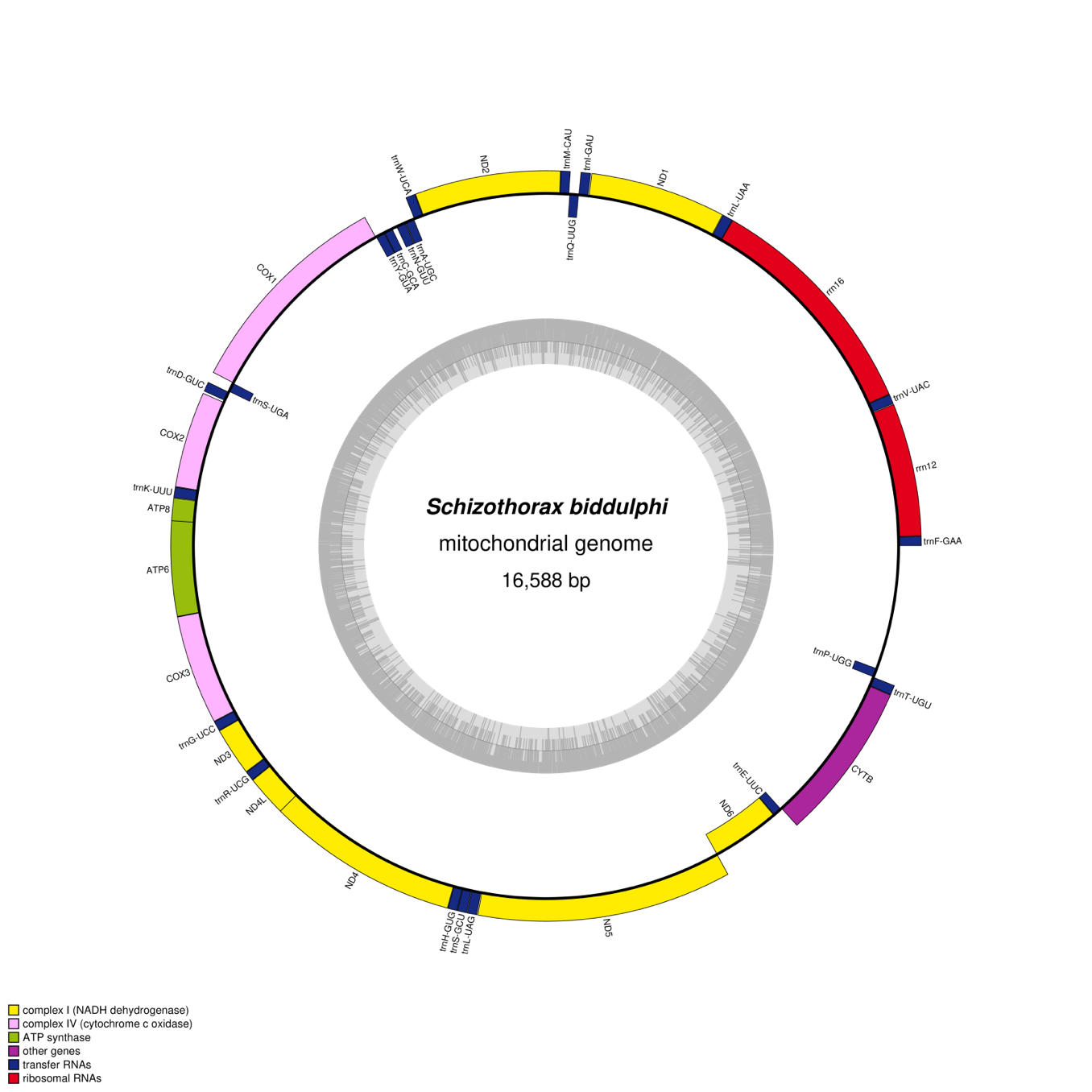
To validate the mitochondrial assembly, we compared it to the reference mitogenomes of related species using BLAST+ and Mitoz.16 The annotated genes in the assembled mitogenome of S. biddulphi showed high identity with the corresponding genes in related species, confirming the accuracy of the assembly. To visualize the organization of the genomic features, circular maps of the S. biddulphi mitochondrial genome were created using OGDraw (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html, accessed on 3 May 2024).17 These maps show the locations of all genes, tRNAs, and rRNAs, offering a detailed view of the mitochondrial structure.
Assessment of Sequence Properties
The nucleotide composition, codon usage, and relative synonymous codon usage (RSCU) of the S. biddulphi mitogenome were analyzed using CodonW.18 This shed light on the nucleotide makeup and codon preferences of the mitogenome. Nucleotide diversity (Pi) and Ka/Ks ratios for the 13 mitochondrial protein-coding genes (PCGs) in Cyprinidae were calculated using DnaSP in order to assess genetic variation patterns.19 Sliding window analyses of the PCGs were also conducted in DnaSP using 100 bp windows with 25 bp steps in order to examine diversity within PCGs. Additionally, genetic distances were estimated using the Kimura-2 parameter (K2P) model in MEGA in order to determine evolutionary relationships. Combining codon usage analysis, Pi, Ka/Ks ratios, and K2P distances enabled us to obtain comprehensive insights into the mitogenomic diversity and evolution of Schizothorax.
Phylogenetic Analyses
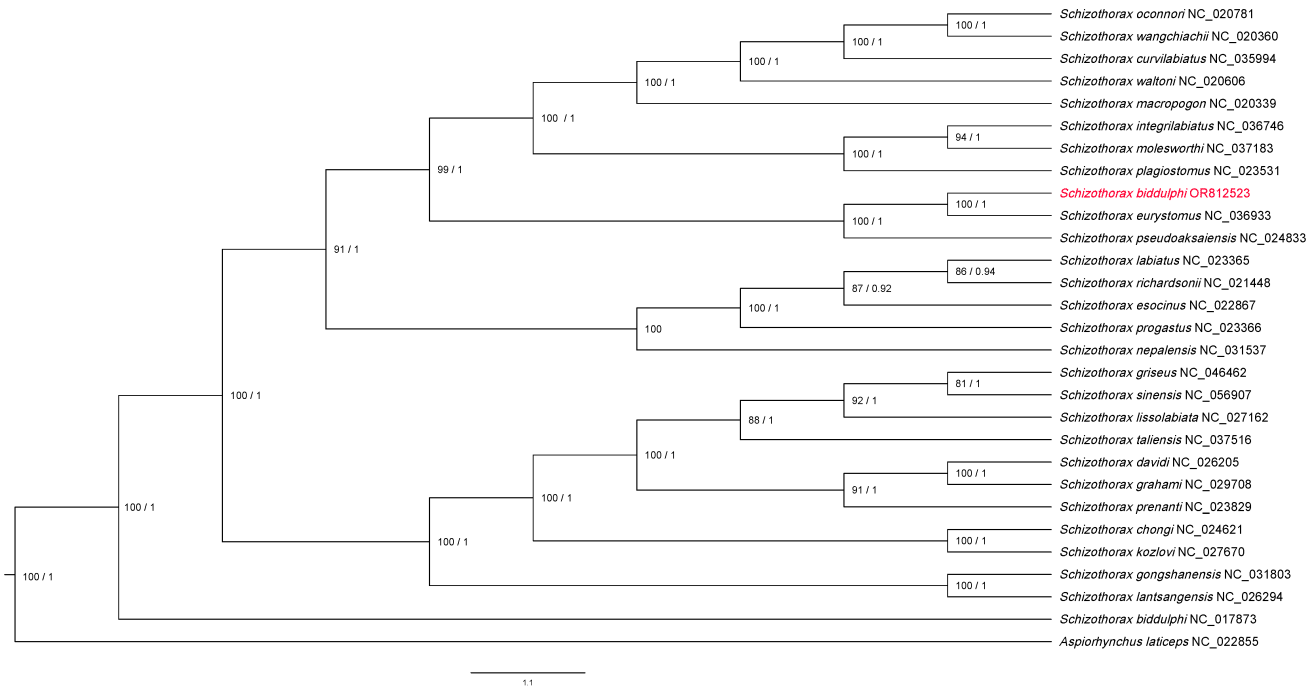
To determine phylogenetic relationships within Schizothorax, the 13 concatenated mitochondrial PCGs from S. biddulphi and other Schizothorax species (Supplement Table 1) were aligned using MAFFT.20 Bayesian inference employed MrBayes with two independent MCMC runs of 200 million generations, sampling every 1000 generations until convergence.21 The first 10% of trees were discarded as burn-in before computing a consensus tree. The bootstraps and posterior probabilities provided statistical support to the evaluation of topology robustness. Bayesian approaches enabled a robust phylogenomic assessment of the evolutionary relationships in Schizothorax to be performed. The phylogenetic tree was visualized with Figtree (v1.4.4), with bootstrap support values and posterior probabilities indicated on the branches.
Results
Genomic Organization and Nucleotide Composition
The mitogenome of S. biddulphi was fully assembled as a 16,558 bp circular molecule (Figure 2), with no gaps or missing regions identified. The assembly was supported by high coverage depth, ranging from 339× to 902×, and 100% of the genome was covered at least 10× (Figure S1). Mapping the HiFi reads back to the assembled genome confirmed uniform coverage across the entire mitogenome, ensuring its completeness. Additionally, the mitogenome was validated by comparing it to known mitochondrial genomes from closely related species, further confirming the absence of any fragmented or missing regions. The average sequencing depth was 607.89×, reinforcing the robustness and accuracy of the assembly. The analysis of its nucleotide composition revealed 28.14% A, 25.64% C, 18.81% G, and 28.41% T, reflecting an AT bias (56.55%) consistent with that of other Cyprinidae species. The structure of the S. biddulphi mitogenome follows the typical organization found in other Schizothorax species, with 13 protein-coding genes, 22 tRNAs, and 2 rRNAs arranged in a typical pattern (Table 1). The shortest tRNAs were tRNAPhe, tRNACys, and tRNASer at 69 bp, while the longest were tRNALeu, tRNAAsn, and tRNALeu at 74 bp. The 896 bp control region lies between tRNAPro and tRNAPhe.

Analysis of Mitochondrial Protein-Coding Genes
The S. biddulphi mitogenome contains a typical set of 28 genes—9 encoded on the L-strand (nad6, trnQ, trnA, trnN, trnC, trnY, trnS, trnE, and trnP) and 19 on the H-strand (atp6, atp8, cox1-3, cob, nad1-5, l-rRNA, s-rRNA, trnD-G, trnH-M, trnR-V, and trnW). The conservation of gene composition and arrangement indicates a shared evolutionary history and suggests that these genes can contribute to phylogenetic resolution at the family level of Cyprinidae species (Figure 3). The observed similarities in gene order and orientation between this study and previous investigations imply the potential utility of these genes in future phylogenetic studies within the family Cyprinidae.

The S. biddulphi mitogenome consists of 3,794 codons across 13 protein-coding genes (Figure 5). Analysis of codon usage reveals important information about gene expression, mRNA stability, and evolutionary relationships. The codon usage in the S. biddulphi mitogenome revealed a strong bias towards specific codons. The most frequent amino acids encoded were Leucine, Serine, Alanine, Arginine, Threonine, and Isoleucine, while Cysteine was the least represented (Table S2). The Ka/Ks ratio (ω) were calculated using the DnaSP software,19 applying the default settings for codon-based analysis. The ratios were interpreted to detect the relative evolutionary pressure on the protein-coding genes. In this study, 12 protein-coding genes showed Ka/Ks values below 1, ranging from 0.012 to 0.147, except for nad1, which had a value of 1.89 (Figure 4). This suggests that purifying selection has significantly influenced most genes, while nad1 has undergone positive selection. Notably, the nad4L and cob genes, which encode components of the respiratory chain—nad4L being part of Complex I and cytochrome b (cob) being part of Complex III—displayed the lowest Ka/Ks ratios (0.012 and 0.018, respectively). These findings indicate stronger evolutionary constraints and a higher degree of functional constraint on these genes. Given that mitochondrial DNA encodes critical components of the respiratory chain and is vital for mitochondrial inheritance, it is particularly prone to the accumulation of deleterious mutations.



The nad2 gene displayed the highest nucleotide diversity with a π value of 0.086, followed by nad6 (0.079), nad1 (0.073), and cob (0.068). Conversely, the genes cox2 (0.034) and atp8 (0.024) showed the lowest nucleotide diversity values. To further explore the genetic distances among these sequences, mean genetic distances were calculated. Reflecting the patterns of nucleotide diversity, nad2, nad1, and cob exhibited higher genetic distances of 0.10, 0.08, and 0.08 respectively, indicating greater sequence divergence. In contrast, cox2, cox3, and atp8 had lower genetic distances of 0.04, 0.04, and 0.03 respectively, suggesting less divergence.
Nucleotide diversity (π) quantifies the average differences between two randomly selected sequences within a gene or genomic region, serving as a critical genetic parameter that measures the level of genetic variation or diversity within a population. Higher π values indicate more diversity in the nucleotide sequences of a specific region, thus allowing researchers to assess the extent of genetic variation present.
This study provide insights into the genetic diversity and sequence divergence in protein-coding genes among Cyprinidae mitogenomes. The identification of genes with high nucleotide diversity and genetic distances, such as nad2, nad1, and cob, suggests that these genes may be subjected to selective pressures or evolutionary forces that contribute to their higher variability. Exploring the functional roles of these genes and their evolutionary implications in Cyprinidae would enhance our understanding of genetic diversity and adaptation in this family.
Phylogenetic Analyses
To ensure robust phylogenetic analysis, our dataset was expanded to 16 mitogenomes. This included 29 from Schizothorax as the focal family and Aspiorhynchus laticeps as outgroups. These reference mitogenomes were retrieved from the NCBI RefSeq database, with data updated as of 17 June 2024.Phylogenetic relationships were investigated using both maximum likelihood (ML). S. biddulphi showed close affinity to Schizothorax eurystomus, in accordance with a prior study performed using morphometric data. Our study provides the first complete Schizothorax phylogenetic analysis, addressing this gap. S. biddulphi’s real taxonomy status provides insights into Schizothoracinae evolution, highlighting the need for further analyses with complementary datasets.
The phylogenetic tree illustrates the evolutionary relationships among various species within the genus Schizothorax and related genera. A. laticeps and S. biddulphi (NC_017873) appear as distinct lineages with minimal genetic distance. The genus Schizothorax is primarily composed of two major clades (Figure 6). The first major clade includes species such as S. biddulphi (this study) and Schizothorax eurystomus, which are closely related with high bootstrap support, along with Schizothorax pseudoaksaiensis. Within this clade, there is a sub-clade consisting of Schizothorax curvilabiatus, Schizothorax oconnori, Schizothorax wangchiachii, and Schizothorax waltoni, all showing high genetic affinity. Additionally, Schizothorax macropogon is part of this group, along with Schizothorax integrilabiatus, Schizothorax molesworthi, and Schizothorax plagiostomus. Another cluster includes Schizothorax esocinus, Schizothorax labiatus, Schizothorax richardsonii, and Schizothorax progastus. Schizothorax nepalensis is a distinct lineage within this clade, indicating significant genetic divergence. The second major clade encompasses species such as Schizothorax chongi and Schizothorax kozlovi, which are highly supported. This clade also includes Schizothorax davidi, Schizothorax grahami, and Schizothorax prenanti. Additionally, Schizothorax griseus and Schizothorax sinensis are closely related to Schizothorax lissolabiata and Schizothorax taliensis. The clade of Schizothorax gongshanensis and Schizothorax lantsangensis is also part of this major group and is highly supported.
Discussion
The characterization of the S. biddulphi mitogenome revealed typical features including AT bias and conserved RNAs and genes, highlighting their functional significance. Variations among Schizothorax species point to a complex interplay between conservation and adaptation. Further investigation of these variations will provide deeper insights into mitogenomic diversity and evolution in Schizothorax. The conservation of gene composition and arrangement indicates a shared evolutionary history and suggests that these genes can contribute to phylogenetic resolution at the family level of Cyprinidae species. The observed similarities in gene order and orientation between this study and previous investigations imply the potential utility of these genes in future phylogenetic studies within the family Cyprinidae.22–31
The Ka/Ks ratios indicate stronger evolutionary constraints and a higher degree of functional constraint on these genes. Given that mitochondrial DNA encodes critical components of the respiratory chain and is vital for mitochondrial inheritance, it is particularly prone to the accumulation of deleterious mutations. The robust purifying selection on the nad4L and cob genes aids in eliminating such mutations, making them ideal molecular markers for phylogenetic analysis. Based on these findings, it is reasonable to infer that these two genes can contribute to the phylogenetic resolution, providing insights into the evolutionary relationships and divergence patterns within this group.32 Genes such as nad6, nad2, nad4, nad5, and cob exhibit higher nucleotide diversity and genetic distances. These genes may be under selective pressures linked to ecological factors such as oxygen availability or temperature fluctuations, potentially contributing to their functional importance in mitochondrial adaptation. These findings are critical for understanding the phylogenetic and conservation dynamics of Schizothorax.
Positive selection on the mitochondrial nad1 gene suggests an adaptive response to ecological or environmental pressures.33,34 As a component of complex I in the mitochondrial respiratory chain, nad1 plays a critical role in cellular energy production by participating in oxidative phosphorylation. The selective advantage of positive selection in this gene may be linked to the organism’s ability to cope with variable environmental stressors such as fluctuating oxygen levels, temperature extremes, or high metabolic demands in specific habitats.33,35 For example, species living in environments where oxygen availability is low or temperature variability is high often exhibit adaptations in mitochondrial function to maintain efficient ATP production. In the case of S. biddulphi, positive selection on nad1 could be associated with metabolic adjustments that optimize energy production under such conditions. Mitochondrial genes, especially those involved in the electron transport chain, are known to undergo rapid adaptation in response to environmental stressors, contributing to an organism’s overall fitness and survival.34,36 Positive selection on the mitochondrial nad1 gene suggests a potential adaptive response, though this signal is preliminary and warrants further investigation in future studies.
Nucleotide diversity provided a critical parameter for quantifying the genetic variation within a population, representing the average differences between two randomly selected sequences within a gene or genomic region.37 Higher π values indicate greater diversity in nucleotide sequences, providing insights into the extent of genetic variation in a given region. Our study revealed that genes such as nad6, nad2, nad4, nad5, and cob exhibit higher nucleotide diversity and genetic distances. These genes may be under selective pressures or evolutionary forces that contribute to their elevated variability. This variability could be linked to functional adaptations or specific ecological pressures acting on the Cyprinidae family. The identification of genes with significant nucleotide diversity highlights important areas for future research into the evolutionary dynamics of Cyprinidae. Understanding the functional roles of these genes, along with their evolutionary implications, will provide deeper insights into genetic diversity and adaptation within this family. This research could have broader implications for conservation efforts and species management strategies. The robust purifying selection on the nad4L and cob genes aids in eliminating such mutations, making them ideal molecular markers for phylogenetic analysis.
Our phylogenetic analysis provides the comprehensive and accurate phylogenetic tree for the Schizothorax genus, revealing important insights into its evolutionary relationships. The high nucleotide diversity observed in certain genes, such as nad1 and nad2, suggests that S. biddulphi may have evolved adaptive traits to environmental changes. These insights could inform conservation strategies by targeting these genes for monitoring and maintaining genetic diversity in the species, particularly in the face of ecological threats such as habitat loss and climate change.
Notably, the mitogenome labeled as S. biddulphi (NC_017873) in the previous study falls outside the principal Schizothorax clades, indicating that this sequence may require further re-examination.13 Within Schizothorax, we also recover two well-supported clades: one comprising S. curvilabiatus, S. oconnori, S. wangchiachii, and S. waltoni, and a second including S. chongi and S. kozlovi. Together, these results support a revised placement of S. biddulphi and underscore the need to re-examine earlier taxonomic assignments using complementary datasets. This study underscores the need for further taxonomic revisions and additional analyses with complementary datasets to address these discrepancies, validating our findings as a more accurate representation of Schizothorax evolution.
Acknowledgments
This work was supported by the Tianshan Talent Training Project of Xinjiang (2023TSYCCX0128),the National Natural Science Foundation of China (31360635), the Corps Science and Technology Bureau Project (2017DB003).
Authors’ Contribution
Conceptualization: Chengxin Wang, Site Luo; Methodology: Linghui Hu, Fangze Zi; Formal analysis and investigation: Huanhuan Wang, Wenxia Cai; Writing - original draft preparation: Chengxin Wang; Supervision: Yong Song, Bin Huo and Xiaotao Shi; Writing - review and editing: Shengao Chen.
Competing of Interest – COPE
The authors declare no conflicts of interest.
Ethical Conduct Approval – IACUC
All experimental protocols were approved by the Science and Technology Ethics Committee of Tarim University (approval code:2023027) and adhered to animal welfare laws, guidelines and policies.
Informed Consent Statement
All authors and institutions have confirmed this manuscript for publication.
Data Availability Statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under accession no OR812523. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA1113635, SAMN41459639 , SRR29086388, respectively.