




Introduction
Prawn (Macrobrachium rosenbergii) recognized as the world’s largest and most economically important freshwater prawn species, is native to the freshwater and brackish water areas of Indo-Pacific region, with its natural distribution encompassing Bangladesh, India, Malaysia, Thailand, and Myanmar.1 Due to its rapid growth and high economic value, it has become one of the top three crustacean species globally. According to the statistics of Food and Agriculture Organization (FAO), the global production of giant freshwater prawn has increased approximately 48-fold since 1980 and reached 337,449.44 tons in 2022, indicating that prawn farming is among the most important industries in the aquaculture. China is the largest prawn producer, accounting for 52.70% of the global total.2 In light of the high demand for prawn products, numerous aquaculture ponds have been used for prawn aquaculture by different means over the past two decades. Limited by culture areas, polyculture is becoming an effective approach to increase prawn production by utilizing the same water resources for two or more compatible species within a single aquaculture season.3,4 Nile tilapia Oreochromis niloticus and common carp Cyprinus carpio are suitable species for prawn due to their distinct feeding habits.5 In current practices, polyculture with different species, such as shrimp Litopenaeus vannamei and crucian Carassius auratus, has become a main practice in prawn aquaculture. However, there has been hardly any effort so far to explore the possibility of polyculture of prawn and shrimp in substrate-based ponds.
Previous studies have demonstrated that probiotic applications can improve growth, feed efficiency, energy transformation, digestive process, water quality maintenance, and resistance to diseases.6,7 The aquaculture industry employs many functional bacteria, including Lactobacillus, Bacillus, Enterococcus, Rhodosporidium, Bifidobacterium, Arthrobacter, Microbacterium, Clostridium, Paenibacillus, Phaeobacter, Roseobacter.8 Some bacteria in aquaculture ponds can remove organic pollutants and decompose feed and feces,9,10 and others, such as Vibrio sp., Flavobacterium sp., Aeromonas sp., and Streptococcus sp., are pathogenic to prawns.11–14 Hence, understanding bacterial community structure is important to evaluate polyculture risks and improve the application of microecology. High-throughput sequencing is a powerful tool to identify bacterial species that could not be recognized by conventional microscopic methods.15,16 Many microbiota of different aquaculture species could be detected by high-throughput sequencing analysis.17–19 The composition of bacterial communities in the intestinal tissue of prawns in traditional ponds and recirculating aquaculture system (RAS) has been investigated.20 An integrated gut microbiota and multi-omics analysis interaction network was constructed, which will aid in the analysis of the growth differences of the female prawns.21 A recent study explored the bacterial community and dominant taxonomies of microbes in the water from polyculture ponds of shrimp and gray mullet Mugil cephalus.22,23 Despite the growing adoption of prawn-shrimp polyculture in practice, critical knowledge gaps persist regarding microbial ecology in pond systems. While previous studies have examined polyculture compatibility in intertidal mudflat ponds,24 limited information is available on the water and sediment bacterial community dynamics and functional interactions in polyculture systems of prawn with shrimp and crucian in the traditional ponds.
This study aims to (1) characterize the bacterial community composition and diversity in water and sediment samples from polyculture systems of prawn with shrimp and crucian using 16S rRNA gene sequencing, (2) compare these microbial profiles with those from monoculture systems to identify polyculture-specific signatures, and (3) evaluate potential linkages between microbial dynamics and ecosystem functions. By integrating high-throughput sequencing with traditional microbiological methods, these findings will provide the first comprehensive assessment of microbial ecology in substrate-based polyculture ponds, offering actionable insights for optimizing sustainable aquaculture practices through microbiome management.
Materials and Methods
EXPERIMENTAL DESIGN AND CULTURE CONDITION
This study was conducted in a prawn commercial farm in the region around Zhejiang province, China. Twelve ponds (area 0.6 ha, 1.2 m depth) were divided into four groups, namely, prawn monoculture (PM), prawn+shrimp (PS), prawn+crucian (PC), and crucian monoculture (CM), 3 replicates per group. A new batch of prawns with a mean initial length of 8.0 ± 0.40 mm was simultaneously introduced into three experimental groups (PM, PS and PC) with an initial density of 600,000 ind ha−1 at the beginning of the experiment. The stocking densities of shrimp (10.0 ± 0.8 mm) and crucian (40.2 ± 8.5 mm) in the PS and PC groups were 150,000 and 9,000 ind ha−1, respectively. The stocking density of crucian (40.2 ± 8.5 mm) in the CM group was 30,000 ind ha−1 (Table 1). Prawns were fed with commercial feed (from 40% to 38% of crude protein according to their growth stages). Daily rations were fed every day with 12% feed per total prawn biomass for the first 30 days. Feeding was later adjusted by observing the feed consumption of prawn, and frequent growth sampling was performed three times a day at 6:00, 12:00, and 17:00. The experiment was conducted for 120 days.
WATER QUALITY SAMPLING AND MEASUREMENT
Temperature (T), pH, and dissolved oxygen (DO) were measured daily in situ 3 replicates per group, by using a hand-held HachHQ40d meter (Hach) at 0.5 m below the water surface. Measurements were taken at the same time each day (8:00 am). The water samples were analyzed for total nitrogen (TN), total phosphorus (TP), nitrite (NO2––N), ammonia (NH4+–N), and chemical oxygen demand (CODMn) at days 15, 30, 60, and 120 of the experiment with 3 replicates per group. Measurements were conducted using the DR3900 spectrophotometer (Hach) with powder pillow procedures, following the detailed protocols outlined in the provided references.
DNA EXTRACTION, PCR AMPLIFICATION, AND HIGH-THROUGHPUT SEQUENCING
At the end of the experiment, bacterial samples from each group were collected from three distinct ponds to account for spatial variability. 5 L of samples of mixed water from 0.5 m below the surface and 1 kg of sediment from the bottom were collected through five-point diagonal-line sampling (positioning sampling points along a diagonal line). Total genomic DNA was extracted using a bacterial genomic DNA extraction kit (Takara, Japan). DNA concentrations and purity were monitored with a Qubit 2.0 Kit. DNA was diluted to 1 ng/μL using sterile water in accordance with the concentration. PCR was conducted in a 30 μL reaction mixture with the following components: 15 μL of 2×Taq master mix, 1 μL of bar-PCR primer F (10 μM), 1 μL of bar-PCR primer R (10 μM), and 10–20 ng of genomic DNA with H2O added to 30 μL. The PCR conditions included an initial denaturation of 3 min at 95 °C, 5 cycles (94 °C for 30 s, 45 °C for 20 s, and 65 °C for 30 s), 20 cycles (94 °C for 20 s, 55 °C for 20 s, and 72 °C for 30 s), and a final extension at 72 °C for 5 min. PCR products from all samples were normalized in equimolar amounts and then sequenced in a concentration of 20 pmol by using an Illumina MiSeq Reagent Kit (500-cycle PE) following the manufacturer’s protocols.
BIOINFORMATIC ANALYSIS
Effective tags were obtained, followed by the splicing of raw data (FLASH, https://sourceforge.net/projects/flashpage/) and cleaning on the chimera (UCHIME, http://drive5.com/usearch/manual/uchime_algo.html). Sequences were then clustered into operational taxonomic units (OTUs) using a 97% similarity threshold.25 Taxonomy was assigned to bacterial OTUs against a subset of Silva database. OTU representative sequences were aligned using PyNAST. The demultiplexed fastq was processed with QIIME2.26 The representative sequence for each OTU was screened for further annotation. In terms of each representative sequence, Greengenes database was used on the basis of the RDP classifier27 algorithm to annotate taxonomic information. The OTU abundance of each sample was generated at the phylum, genus, class, order, and family levels using the QIIME pipeline. The core bacteria and potential pathogenic microbial taxa were also selected. The abundance and diversity indices were generated using mothur with an OTU identity cutoff of 97% after implementing a pseudo-single linkage algorithm. The alpha diversity richness and evenness of the bacterial community were calculated using Chao1, ACE, Shannon, Simpson, and Coverage. Hierarchical cluster analysis was performed using the Bray–Curtis similarity based on the abundance of all OTUs on R software with the statistical package vegan. Nonmetric multidimensional scaling (NMDS) was conducted to investigate the differences in bacterial community structure among samples on the basis of the Bray–Curtis dissimilarity by using standardized and square-root transformed data. Analysis of similarities was used to test statistically whether a significant difference exists among models. Differentially abundant microbial taxa were detected for each model with a linear discriminant analysis effect size (LEfSe)28 by using a cutoff of LDA > 2 and P < 0.05 for the internal Kruskal-Wallis and Wilcoxon tests.
DATA ANALYSIS
Data analysis was performed with SPSS 17.0. The results were presented as mean ± standard error. Statistical differences were measured and compared among different groups using one-way ANOVA, followed by the Duncan LSD method for all-pairwise comparison to identify specific group differences. P value at < 0.05 was used to indicate statistical significance. GraphPad Prism (version 7) was used to conduct statistical tests and graphs.
Results
WATER QUALITY IN DIFFERENT CULTURE GROUPS
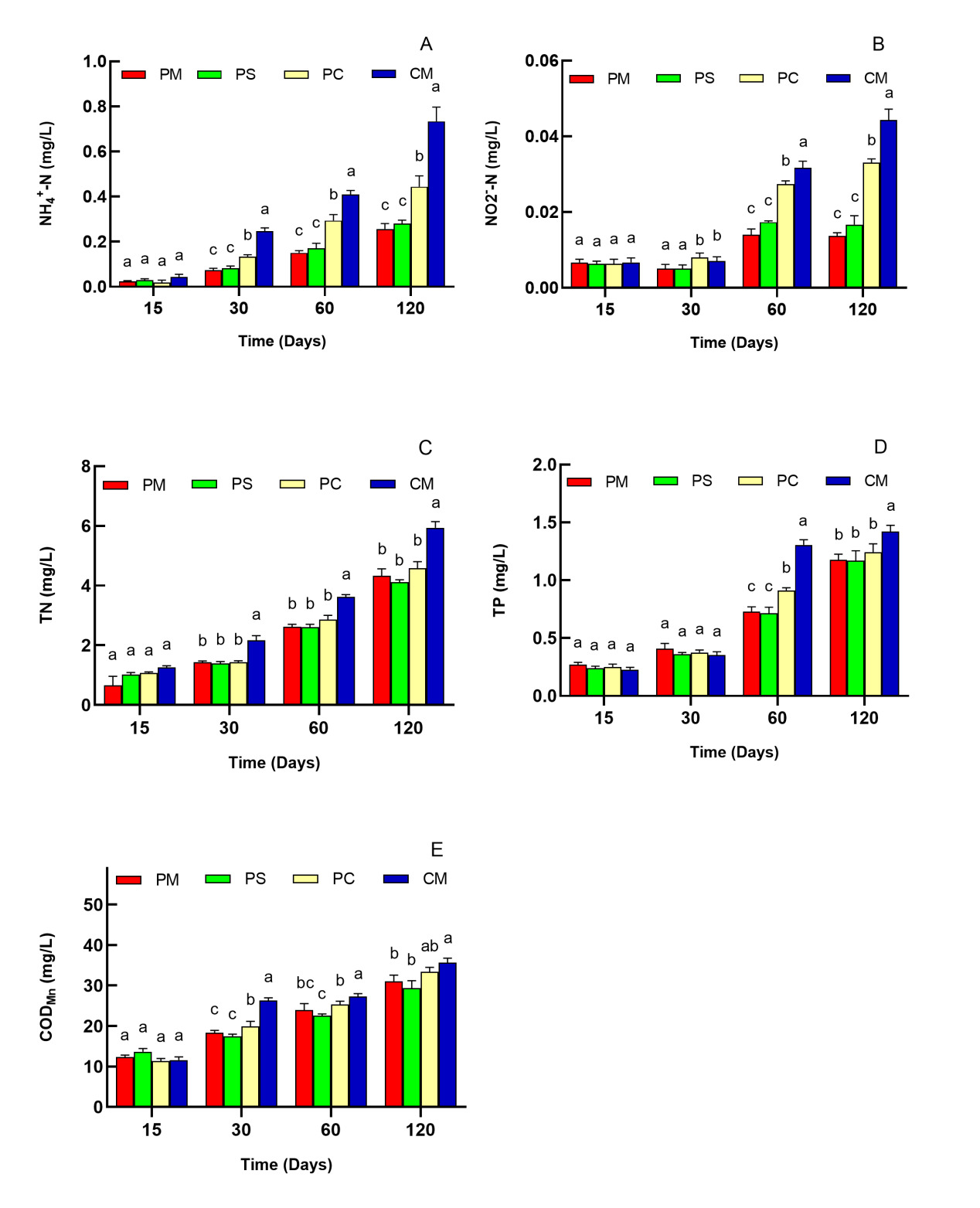
Water temperature, pH, and DO were detected daily. DO remained at >5.0 mg/L, whereas pH ranged from 7.4 to 8.5, which were suitable for the growth of cultured animals in this study. Temperature changed gradually with season and time, but no significant difference was found among the different culture systems. Thus, the three parameters were not demonstrated in this research. Fig. 1 illustrates the variations in NH4+–N, NO2-–N, TN, TP, and CODMn concentrations among different systems. All of the parameters increased with time. NH4+–N concentration in PS group was in accordance with PM group, but PC and CM groups showed significant increase from 30 days to 120 days (P<0.05, Fig. 1A). The NO2-–N concentrations were consistent with NH4+–N, (Fig. 1B). TN concentrations in polyculture groups (PS group and PC group) showed no significant difference with the PM group (P>0.05) (Fig. 1C). TP concentrations in CM and PC groups increased at 60 days, while there was no significant difference between PM and PS groups throughout the whole experiment (Fig. 1D). The concentration of CODMn also exhibited significant increase in the CM group from 30 to 120 days (Fig. 1E).
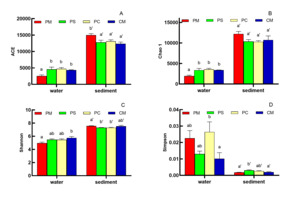
DIVERSITY AND ABUNDANCE OF BACTERIA IN DIFFERENT CULTURE GROUPS
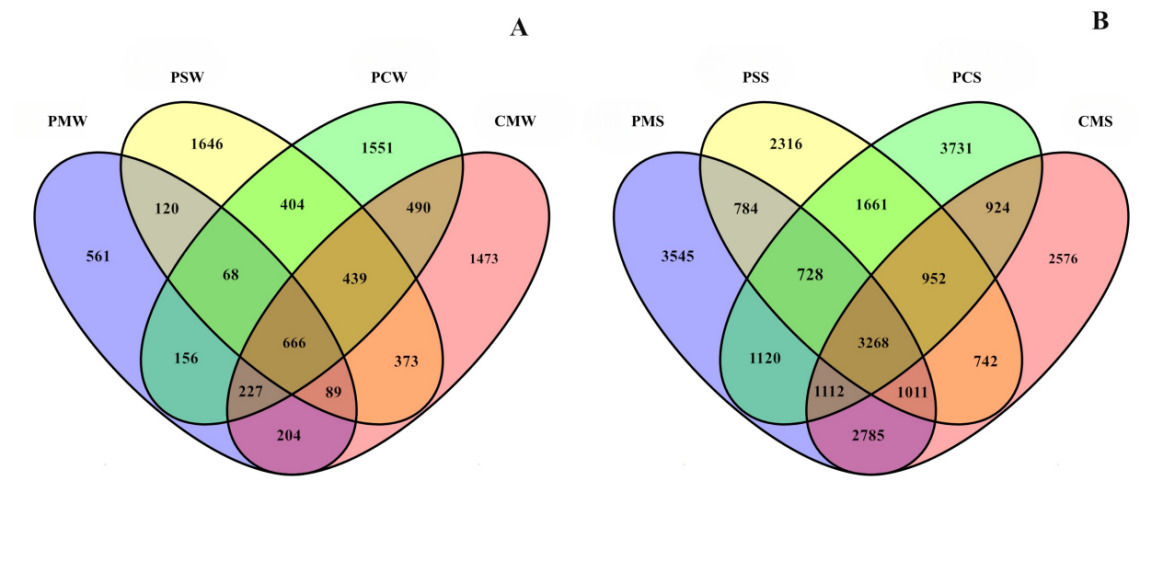
A total of 785219 raw sequences, including 1600 chimeras, 160 out-targets, 335175 organelles, and 448284 filtered sequences, from 12 samples (3 samples for each group) from the water of different culture groups were obtained after the Illumina sequencing of the V3–V4 segments of the 16s rRNA gene. In sediment, a total of 708925 sequences, including 3597 chimeras, 183 out-targets, 24491 organelles, and 680654 filtered sequences, were obtained for sediment samples after the Illumina sequencing of the V3–V4 segments of the 16s rRNA gene (Table 2). Sequences with ≥97% similarity were assigned to the same OTUs for further annotation. The average OTUs of different culture groups (PM, PS, PC, and CM) were 2091, 3961, 3805, and 4001 (Fig. 2A), whereas the average OTUs in sediment of different culture groups were significantly higher (PMS = 14262, PSS = 13370, PCS = 11462, and CMS = 13496) than those in water (Fig. 2B).
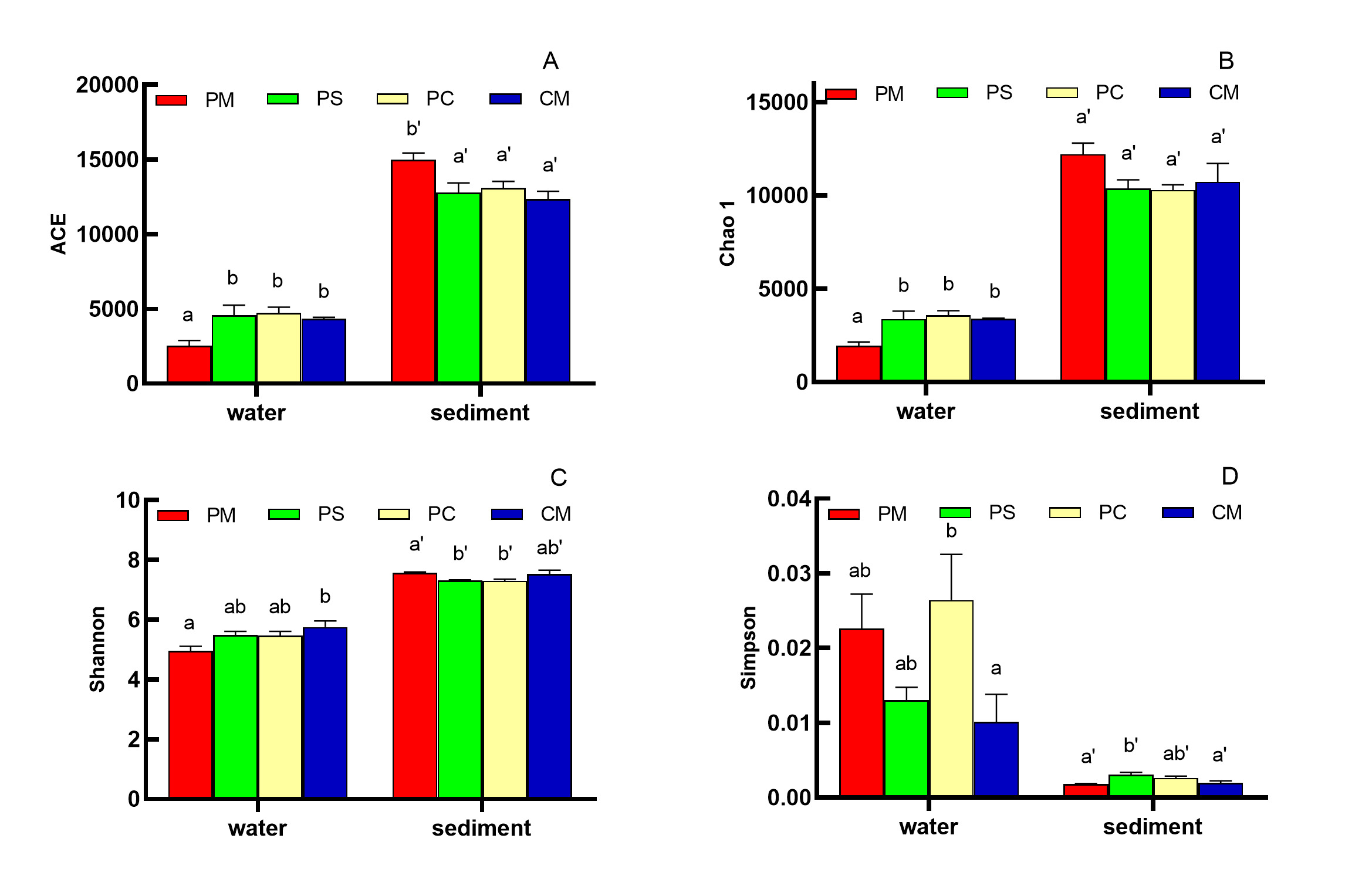
The alpha diversities of bacteria among the different culture groups were also analyzed (Fig. 3). The observed species rarefaction curves (not shown) reflected that the sequence data sufficiently produced stable and unbiased estimates of species richness. The alpha diversity indices showed that the ACE indices of PS, PC and CM group in water was significant higher than that in PM group, whereas the ACE indices in sediment reached the highest level in the PM group (P < 0.05, Fig. 3A). Chao 1 and Shannon indices revealed similar results to that of ACE (Fig. 3B, Fig. 3C). On the contrary, the Simpson indices in water recorded the highest result in the PC group, whereas the Simpson indices in sediment demonstrated the highest result in the PS group (Fig. 3D). The ACE, Chao 1, and Shannon indices in sediment were significantly higher than those in water. However, the Simpson indices suggested opposite results.
COMPARISON OF BACTERIAL COMMUNITY STRUCTURE AMONG DIFFERENT CULTURE GROUPS
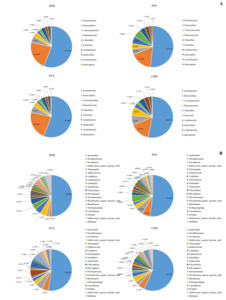
The bacterial community structure in water among different culture groups are shown in Fig. 4. At the phylum level, the polyculture groups shared the same dominant phyla with one another. Proteobacteria were the most dominant phyla, occupying 34.98%–45.01% of the water in different culture groups. Bacteroidetes, Verrucomicrobia, Actinobacteria, Planctomycetes, and Chloroflexi were the other dominant phyla in different culture groups (Fig. 4A).
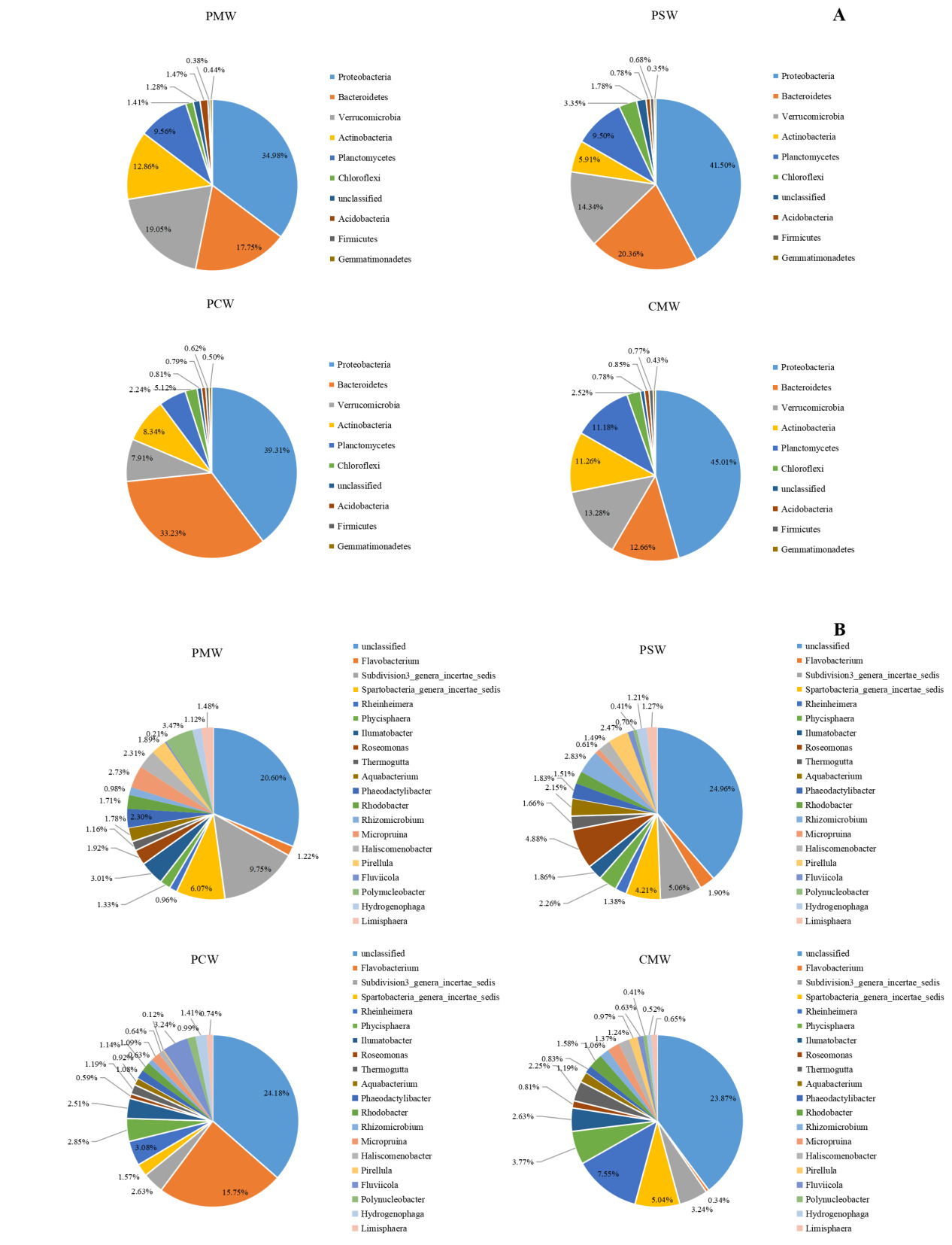
At the genus level, 194.67 ± 15.94, 326.67 ± 21.40, 343.67 ± 16.70, and 335.00 ± 27.50 genera that belonged to 22.00 ± 2.08, 33.33 ± 2.33, 25.33 ± 1.67, and 24.33 ± 1.67 phyla were identified from PM, PS, PC, and CM, respectively. A total of 20.60% ± 0.85%, 24.96% ± 2.14%, 24.18% ± 1.37%, and 23.87% ± 4.07% unclassified species were found in the PM, PS, PC, and CM groups, respectively. The bacteria in water were mainly aerobic bacteria. Spartobacteria_genera_incertae_sedis, Phycisphaera, Ilumatobacter, Thermogutta, and Rhodobacter were the main common abundant species in different culture groups. Roseomonas, Phaeodactylibacter, and Pirellula were abundant in the PM and PS groups. Micropruina, Haliscomenobacter, and Polynucleobacter were the only prevailing abundant genera in the PM group. Aquabacterium and Rhizomicrobium were the only abundant genera in the PS group. The PC group recorded the highest genera of Flavobacterium, Rheinheimera, and Fluviicola. Litorilinea was the most abundant genus in the CM group (Fig. 4B).
The bacterial community structure in sediment among different culture groups are shown in Fig. 5. Similar to the aforementioned results of water, the polyculture groups shared the same dominant phyla with one another in sediment at the phylum level. Proteobacteria, Bacteroidetes, Verrucomicrobia, Actinobacteria, Planctomycetes, and Chloroflexi were the dominant phyla in different culture groups. Actinobacteria and Spirochaetes were the other dominant phyla that were not dominant in the water of different culture groups (Fig. 5A).
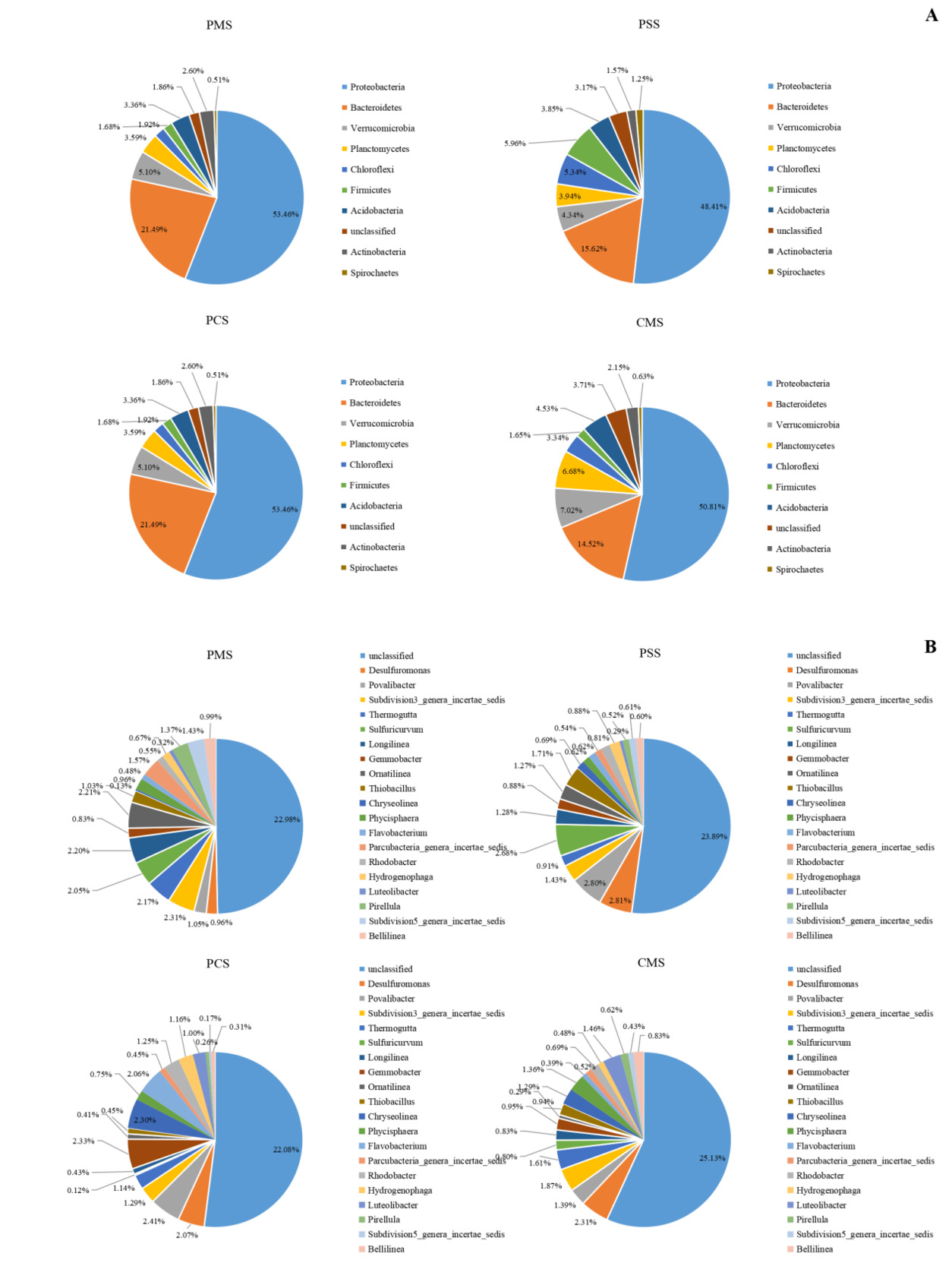
At the genus level, species changed significantly among different culture groups. Various species (697.33 ± 14.50, 681.00 ± 19.92, 709.67 ± 18.48, and 708.00 ± 19.29) were detected from 46.00 ± 0.58, 45.00 ± 1.73, 41.33 ± 1.33, and 42.33 ± 0.33 phyla in PM, PS, PC, and CM, respectively. A total of 22.98% ± 1.92%, 23.89% ± 2.02%, 22.08% ± 0.46%, and 25.13% ± 1.89% unclassified species were found in the PM, PS, PC, and CM groups, respectively. Only four species (Desulfuromonas, Povalibacter, and Thermogutta) were abundant in all groups. The PM and PS groups also shared the same dominant genera of Sulfuricurvum, Longilinea, Ornatilinea, and Thiobacillus. Gemmobacter, Flavobacterium, Rhodobacter, Hydrogenophaga, and Azoarcus were the only prevailing abundant genera in the PC group, whereas Phycisphaera, Luteolibacter, and Thioprofundum were the only abundant genera in the CM group. In addition, many anaerobic bacteria (Desulfopila, Desulfobacteraceae, Desulfobulbaceae, Desulfuromonadaceae, and Desulfococcu) were detected in sediment among different culture groups (Fig. 5B).
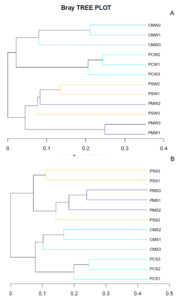
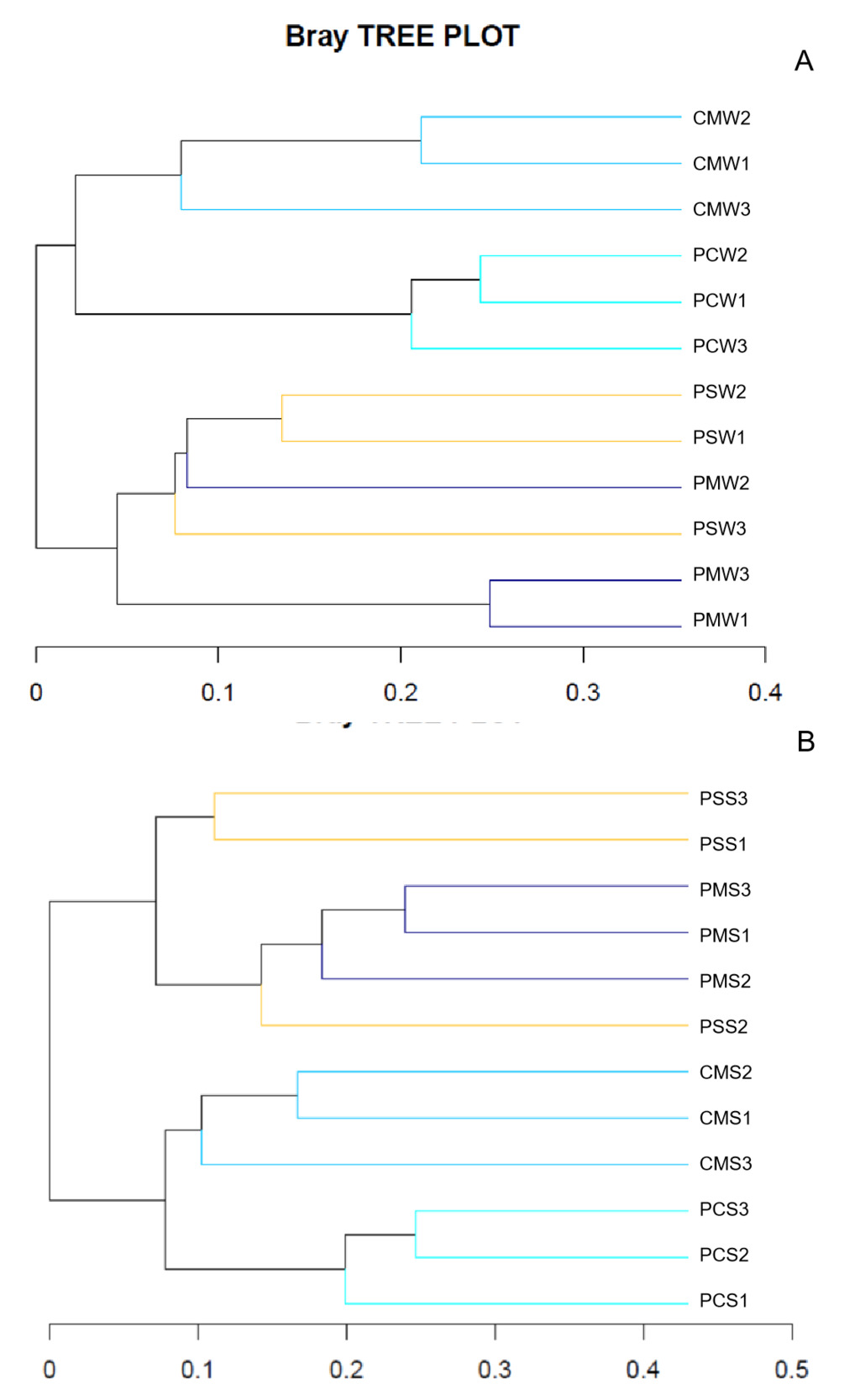
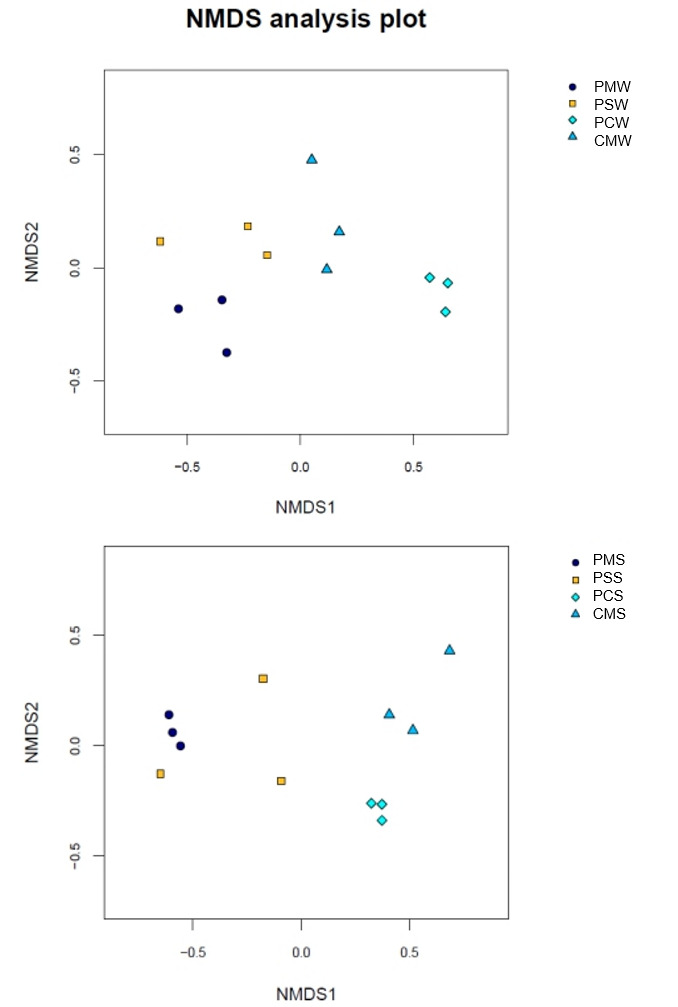
A hierarchical cluster tree based on Bray–Curtis was constructed in accordance with the OTU distribution to illustrate the microbial community of different culture groups. The PM and PS culture groups were grouped together, whereas the PC and CM culture groups were grouped together (Fig. 6A). In sediment, the PM and PS culture groups were also grouped together (Fig. 6B). The NMDS also showed a differentiated distribution of microbial taxa and presented similar results to those of the hierarchical cluster tree (Fig. 7).
LEfSe confirmed that each treatment had its own indicator taxa, from phylum to genus. Intergroup comparison was conducted using LEfSe (LDA > 2.0, P < 0.05) to identify species with significant changes under different treatments (Fig. 8). In total, 45 bacterial clades presented a significant difference in water among the different culture systems, i.e., 24 for the PM group, 3 for the PS group, 7 for the PC group, and 11 for the CM group. The potential biomarkers decreased in the polyculture systems (PS and PC groups). Roseomonas, Methylovorus, Algoriphagus, Acidothermus, Flavobacterium, and Hyphomonas were the main genera among different culture systems (Fig. 8A). For sediment, 69 clades with significant difference were found, i.e., 27 for the PM group, 7 for the PS group, 9 for the PC group, and 26 for the CM group. Syntrophorhabdus, Mangrovibacterium, Azoarcus, Defluviimonas, and Cetobacterium were the main genera among different culture systems. Polyculture systems also decreased the potential biomarkers (Fig. 8B).
_effect_size_taxonomic_cladograms_comparing_the_samples_.png)
POTENTIAL PATHOGENS IN THE WATER OF DIFFERENT CULTURE GROUPS
Differences were revealed in the potential pathogens in water among different culture groups. The common pathogens, including Aeromonas, Clostridium, Flavobacterium, Heliobacillus, Lachnospira, Leucothrix, Mycobacterium, Plesiomonas, Providencia, Pseudomonas, Rickettsia, Thiothrix, and Vibrio, of prawn and other aquaculture species were calculated. Flavobacterium and Roseomonas were the main components of the total potential pathogens. The PC group recorded the highest levels of Flavobacterium, Mycobacterium, and total potential pathogens compared with other groups (P < 0.05). The Pseudomonas in the CM group was significantly higher than those in other groups. Nevertheless, none of the potential pathogens reached the peak in the PM group. Although differences were observed among different culture groups, the reads of many bacterial species, such as Flexibacter, Heliobacillus, Lachnospiracea_incertae_sedis, Leucothrix, NHPB, Plesiomonas, Providencia, Thiothrix, Vibrio, Thiopseudomonas, and Piscirickettsia, were maintained in a very low level or not detected in the ponds of different culture groups (Table 3). There were also differences in the potential pathogens in sediment among different culture groups. Similar to the results in water, most potential pathogens species, namely, Flexibacter, Heliobacillus, Lachnospiracea_incertae_sedis, Leucothrix, NHPB, Plesiomonas, Providencia, Rickettsia, Thiothrix, Vibrio, Diplorickettsia, and Piscirickettsia, were undetected or observed at extremely low levels. The PC group recorded the highest levels of total potential pathogens. The levels of Flavobacterium and Mycobacterium in the PC group were significantly higher than those in the other groups. Thiopseudomonas and Diplorickettsia also demonstrated a significant increase in the PC group. The CM group presented the highest level of Pseudomonas. However, different from the results in water, the PM group showed considerable levels of Aeromonas and Clostridium sensu stricto (Table 4).
Discussion
The aquaculture environment plays a vital role in aquatic organism production.29,30 Among the key water quality parameters, ammonia nitrogen accumulation can disrupt enzymatic functions and compromise osmotic balance,31 while nitrite nitrogen serves as a crucial indicator throughout the whole aquaculture process.32 Thus, monitoring and assessment of the water quality can aid in identifying the critical conditions for farming and controlling the harmful risks of environmental impairment in the whole crustacean culture production.33 In this study, significantly lower concentrations of key parameters (NH4+–N, NO2-–N, TN, TP, and CODMn) were observed in PM and PS groups compared to the PC and CM groups at the mid-late period of the experiment. These differential patterns likely reflect fundamental physiological distinctions between crustaceans and fish species within the aquaculture environment.
Healthy bacterial communities serve as sensitive environmental bioindicators, reflecting the overall aquaculture condition.34 As fundamental components of the culture environments, water and sediment play important roles in the growth of crustaceans through their physicochemical and biological interactions.35,36 Distinct environmental factors may shape the bacterial communities in the crustacean intestine and the surroundings.37 Water and sediment are important locations for matter exchange in an aquaculture pond.35 Bacterial communities of the water and sediment were more complex than those of the gastrointestinal tract.38 Thus, monitoring and assessing the bacterial communities of ponds can help understand the changes in bacterial diversity and composition and control the harmful risks of environmental impairment. In this study, the average OTUs of polyculture systems were significantly increased than that in PM group in water. The substantially higher number of unique OTUs in the polyculture systems suggests that polyculture provided a more favorable niche for microbial colonization, consequently enhancing α-diversity." Moreover, the bacterial communities of four different culture systems only shared 666 and 3268 OTUs. These findings highlight the dynamic interplay between aquaculture environments and microbial ecosystems. The observed variations in OTUs across different culture systems may be attributed to host-microbe-environment tripartite interactions.
ACE and Chao 1 indices can refer to the total abundance-based coverage in a community, whereas Shannon–Wiener diversity indices can indicate species richness and evenness, which can reflect the species diversity of the samples objectively.39 Notably, the microbiota of water and sediment in polyculture groups displayed significantly higher ACE, Chao 1 and Shannon indices, reflecting a more diverse microbial composition. Such diversity may facilitate a broader array of metabolic pathways and enhance immune resilience, potentially contributing to better disease resistance and overall health. Previous investigations have documented the diverse alpha diversity of bacterial communities in shrimp ponds.7,40 On the contrary, Abraham41 also recorded a marked reduction in luminous bacterial abundance in a polyculture pond of shrimp and fish. Different alpha diversity of bacterial community among different culture systems and environments, suggesting a culture system-dependent microbial community structure. Bacterial diversity is generally higher in sediment than in water among different aquaculture systems, which might be attributed to the substrate-specific selection pressures, as sediment’s anoxic conditions favor anaerobic taxa while the water column selects for aerobic species.. These patterns underscore the intricate tripartite interactions between culture systems, environmental parameters, and microbial community assembly.
Our results reveal striking similarities in bacterial communities at the phylum level among different culture systems, with Proteobacteria dominating in all culture groups. Similar results have been reported in many previous studies.42,43 LEfSe analysis revealed significant differential abundance of Proteobacteria classes (Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, and Deltaproteobacteria) among different culture groups in water and sediment, reflecting their different functions in the aquaculture system.44 Bacteroidetes was the most numerous phylum in the samples with nitrifying and denitrifying biofilters.45 Many species of Bacteroidetes have been proven to be good degraders of organic matters.46,47 Sun reported that Bacteroidetes was one of the most abundant phylum in the water and sediment of Eriocheir sinensis, corroborating its ecological significance in aquatic systems.48 The PC group had abundant Bacteroidetes on the basis of the LEfSe analysis, which was probably due to the adequate substances in this group. However, Bacteroidetes was also abundant in the sediment of the PM group. The reasons for this discrepancy might be the deposit increases with the long-term culture history, other than the culture model.49 Cyanobacteria are the most common phytoplankton, and their abundance has been closely associated with the influx of nutrients. Abundant Cyanobacteria have been reported in many former studies.50,51 On the contrary, cyanobacteria were scarcely detected in the waters and sediments in this study. This unexpected absence challenges conventional assumptions about polyculture systems as potential bloom incubators, suggesting alternative microbial regulation mechanisms may be operating. The detection of Fibrobacteres in this study provided compelling evidence for active cellulose degradation pathways, particularly in nutrient-rich aquaculture environments where synergistic microbial interactions likely facilitate complex organic matter decomposition.52 At the genus level, a significant increase in genus was detected in polyculture systems. This result was consistent with the results of alpha diversities. Polyculture practices obviously altered microbial community composition. Specifically, the PS group exhibited a marked increase in the proportion of Roseomonas and Phaeodactylibacter, which are critical for participating in denitrification and reducing nitrate levels. Unclassified genera were the most abundant in the waters and sediments, indicating the existence of numerous bacterial species without annotations. This study revealed that the bacteria in aquaculture water were mainly aerobic bacteria, whereas the bacteria in aquaculture sediment were mainly aerobic bacteria. These phenomena were related to the different dissolved oxygen environments.
Bacterial communities had a close relationship with cultured animals, which varied in different aquaculture environments.53 In this study, PM and PS culture groups were grouped together by A hierarchical cluster tree based on Bray–Curtis. This result clearly indicated the crucial role of culture species, especially their genetic relationship, in shaping bacterial communities. LEfSe confirmed that each treatment had its own indicator taxa, from phylum to genus, demonstrating that aquaculture practices profoundly reshape microbiome structure. Notably, polyculture systems exhibited markedly fewer potential biomarkers than monocultures both in water and sediment. This reduction suggests enhanced microbial stability under polyculture conditions, corroborating studies where diversified systems suppress dominant taxa through competitive exclusion and niche partitioning.
Imbalances in microbial compositions triggered by poor water quality will lead to an increase in potential pathogens, which further compromise cultured animals’ health.17 Aeromonas, Escherichia, Flavobacterium, Pseudomonas, Rickettsia, and Vibrio are the main pathogenic bacteria of fish and crustacean.24,54 Various potential pathogens were detected in this study, meaning the aquaculture surroundings were complex and critical to the growth and rearing of cultured animals.55 Vibrio, a gram-negative halophilic bacterium, causes severe devastation in prawn culture and often dominates other bacterial species.56,57 Aeromonas, known as one of the most common pathogens, exhibited devastating effects on the survival rates and production of aquaculture species.58,59 However, Vibrio and Aeromonas levels remained very low across all culture systems in this study. Flavobacterium is also an important opportunistic pathogenic species.60 In this study, Flavobacterium was the highest potential pathogen both in the water and sediment. In addition, Flavobacterium in the PC group was much larger than those in any other groups, both in water and sediment. This finding was corroborated by LefSe analysis, which revealed a high LDA score for Flavobacterium in the PC group. Total potential pathogens followed the same pattern as that of Flavobacterium, implying the presence of disease risk in the PC group.
Conclusion
This study revealed the water quality and changes in the bacterial communities in water and sediment in response to the system of monoculture and polyculture. PS group maintains good water quality. Significant increases in alpha diversity, phylum, and genus were also detected in polyculture systems. Many potential pathogens persisted at negligible levels across all culture systems. However, total potential pathogens in the PS group were in accordance with the PM group, while the PC group revealed a significant increase of potential pathogens. The shift of bacterial communities was related to the genetic relationship between cultured animals. These results demonstrate that the prawn-shrimp polyculture system not only maintains stable water quality but also minimizes pathogenic risks, supporting its feasibility as an ecologically balanced aquaculture strategy.
Acknowledgments
This study was financially supported by The Basic Public Welfare Program of Zhejiang Province (LGF22C190001).
Authors’ Contribution
Conceptualization: Meng Ni (Lead). Methodology: Meng Ni (Equal), Julin Yuan (Equal). Formal Analysis: Meng Ni (Equal), Songbao Zou (Equal), Mei Liu (Equal), Dan Zhou (Equal). Investigation: Meng Ni (Equal), Songbao Zou (Equal), Mei Liu (Equal), Dan Zhou (Equal). Writing – original draft: Meng Ni (Lead). Writing – review & editing: Meng Ni (Equal), Julin Yuan (Equal). Funding acquisition: Meng Ni (Equal), Julin Yuan (Equal). Resources: Meng Ni (Equal), Songbao Zou (Equal), Mei Liu (Equal). Supervision: Meng Ni (Equal), Dan Zhou (Equal).
Competing of Interest – COPE
Competing interests’ statement: No competing interests were disclosed. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical Conduct Approval – IACUC
The sample collection and experiments in the study were in compliance with the Animal Ethics Committee of Zhejiang Institute of Freshwater Fisheries (Animal Ethics No. 1067, March 6, 2019).
Informed Consent Statement
All authors and institutions have confirmed this manuscript for publication.
Data Availability Statement
All are available upon reasonable request.