1. Introduction
Atrina pectinata belongs to the family Pinnidae and is naturally distributed along the Indo-West Pacific coast, ranging from south-eastern Africa to Melanesia and New Zealand. In China, populations extend from the Liaodong Peninsula in the north to the Qiongzhou Strait in the south.1,2 As a commercially valuable bivalve in East Asia (especially China, Korea and Japan), its dried adductor muscle is esteemed as one of the “Eight Treasures of the Sea” and market demand continues to rise.3 However, over-exploitation has severely depleted wild stocks in Chinese waters, and natural harvests can no longer meet domestic and international demand. Since the early 1980s, China has conducted artificial breeding research on A. pectinata, laying the foundation for aquaculture technology.1,4 Nevertheless, seed-rearing techniques remain imperfect, and selective breeding has lagged behind that of other economically important bivalves, such as oysters and scallops. Without molecular breeding data, traditional propagation suffers from low larval survival, severe larval adhesion, and difficulty in inducing spawning, all of which constrain large-scale seed production.5 Elucidating the molecular mechanisms governing sex determination and gonadal development in A. pectinata is, therefore, both a fundamental biological need and a key step toward overcoming aquaculture bottlenecks and achieving sustainable resource use.
In recent years, significant advances have been made in understanding the molecular regulation of sex determination and gonadal development in bivalves. Key transcriptional regulators, including Dmrt1/2, Foxl2, Sox, β-catenin, Fem-1, CYP, 17β-HSD, Nanos, and Hsp70 have been identified.6–12 These factors belong to different regulatory modules that collectively maintain or modulate bivalve sex determination and gonadal developmental rhythms: the Wnt/β-catenin-Foxl2 axis drives ovarian differentiation, the Dmrt1-Sox9 axis governs testis development,13–15 the DMRT1–FEM-1 ubiquitin-stabilization loop16 and Nanos–β-catenin germ-cell–somatic-cell crosstalk17 participate in dosage regulation, 17β-HSD and CYP are involved in sex-steroid synthesis,18–20 and Hsp70 mediates external-signal processing (stress & energy sensing). However, the functional conservation and specificity of these mechanisms in A. pectinata remain unclear, and molecular markers, key pathways, and regulatory networks underlying gonadal development in this species are virtually absent, making it difficult to assess the transferability of findings from other bivalves. In particular, A. pectinata possesses a unique elongated shell, a deep-buried life history, and a prolonged sexual maturation period, biological characteristics that may render its spatiotemporal pattern of gonadal development molecularly distinct from other bivalves.
We hypothesize that the molecular mechanisms of sex determination and gonadal development in A. pectinata are evolutionarily conserved yet exhibit species-specific features. To test this hypothesis, we collected gonadal samples at different developmental stages, performed histological sectioning to characterize gonadal development, and employed high-throughput transcriptome sequencing combined with bioinformatics to construct the gene-expression profile during peak gonadal development. Key differentially expressed genes (DEGs) were screened to preliminarily demonstrate the conservation and specificity of the molecular regulatory network. Our findings will provide critical insights into sex- and reproduction-related molecular regulation in A. pectinata, offer candidate genes for in-depth mechanistic studies, and lay a theoretical foundation for optimizing artificial breeding strategies.
2. Materials and Methods
2.1. Sample Collection and Gonadal Histology
Wild two-year-old A. pectinata were collected from Changdao, Yantai, China (37°55′47.0748″N, 120°45′35.6148″E) on April 28, May 21, June 28, and July 19, 2024. At each sampling time point, three individuals of each sex were collected. For histological examination, gonadal tissues were immediately dissected and fixed in Bouin’s solution. Fixed tissues were processed through a graded ethanol series for dehydration, cleared in xylene, and embedded in paraffin. Sections (4.5 μm thickness) were cut using a microtome, dewaxed, rehydrated through a graded ethanol series, and stained with hematoxylin and eosin (HE). Stained sections were examined microscopically and photographed to document gonadal morphology.
2.2. High-Throughput RNA Sequencing
For transcriptome sequencing, gonadal samples (mature stage) collected on June 28, 2024, were immediately frozen in liquid nitrogen. Three female gonadal samples were designated as FX1, FX2, and FX3, while three male samples were designated as MX1, MX2, and MX3. High-throughput RNA sequencing was performed by Shanghai Shenggong Biological Co., Ltd, and a reference-free (de novo) transcriptome analysis strategy was employed. Total RNA was extracted using TRIzol reagent, treated with DNase I to remove genomic DNA, and then verified by agarose gel electrophoresis to ensure the absence of visible genomic DNA bands. RNA quality was confirmed by an RNA Integrity Number > 7.0, and an A260/A280 ratio of approximately 2.0, with a total RNA yield ≥5 μg in all samples. The cDNA library was constructed using the Hieff NGS™ MaxUp Dual-mode mRNA Library Prep Kit. Throughout the experiment, all samples were processed under a strictly unified protocol, and libraries were prepared simultaneously to minimize batch-effect interference. After quality control and assessment, sequencing was carried out on the BGI Genomics DNBSEQ-T7 high-throughput sequencing platform.
2.3. Bioinformatics Analysis
Raw sequencing reads were quality-assessed using FastQC, followed by adapter removal and quality trimming using Trimmomatic. Clean reads were assembled using Trinity to generate unigenes, and redundant sequences were removed. The assembled unigenes were functionally annotated using multiple databases, including Conserved Domain Database (CDD), Eukaryotic Orthologous Groups (KOG), Clusters of Orthologous Groups (COG), Non-redundant Protein Sequence Database (NR), Nucleotide Sequence Database (NT), Protein Family Database (PFAM), and Gene Ontology (GO). Differential expression analysis was performed using DESeq with thresholds of q-value < 0.05 and |fold change (FC)| > 2. GO enrichment analysis was conducted using top GO, while Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using clusterProfiler.
2.4. Quantitative real-time PCR (RT-qPCR) Verification
To validate the RNA-seq results, three DEGs (β-catenin, spata6, and sox2) were randomly selected for RT-qPCR analysis, using 18S rRNA as the internal reference gene. The relative expression levels were calculated using the 2-ΔΔCt method and presented as log2(FC). For each group, three independent biological replicates were analyzed. Statistical significance of the expression differences between the two groups was determined using an independent samples Student’s t-test. Data visualization was performed using Origin 2021 software.
3. Results
3.1. Observation of Gonadal Paraffin Sections
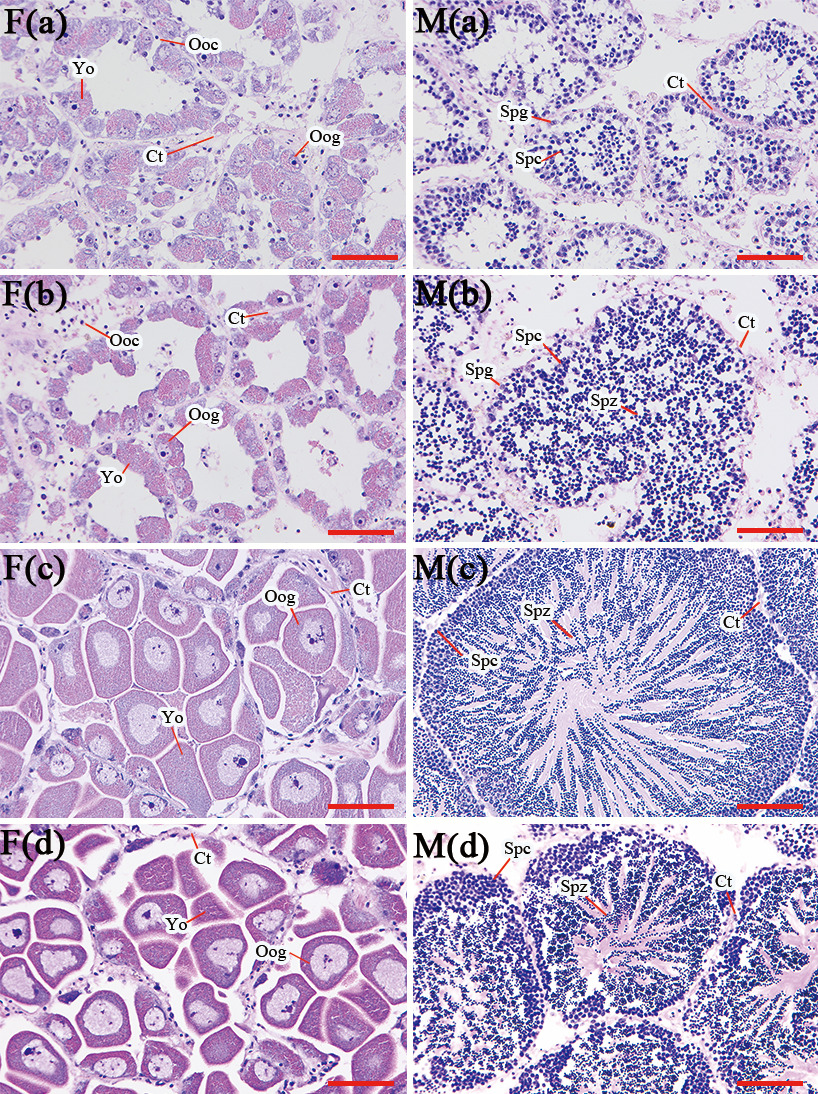
Histological examination revealed distinct stages of gametogenesis in both male and female A. pectinata throughout the sampling period (Fig. 1). On April 28, the follicles in both male gonads (M(a)) and female gonads (F(a)) were small, with a visible cavity in the center of the follicles. The cells within these follicles were primarily spermatogonia and spermatocytes in males, and oogonia and early-stage oocytes in females, indicating that the gonads were at early stages of gametogenesis. By May 21, the female gonad (F(b)) remained in the early stages of gametogenesis, whereas the male gonad (M(b)) exhibited some follicles containing spermatozoa, a result of meiotic division, suggesting the gonads were at the mid-stage of gametogenesis. On June 28, the female gonad (F(c)) displayed fully developed follicles with a few oogonia, while a large number of oogonia were observed to be fully developed, tightly packed, and irregularly shaped. In the male gonad (M(c)), a small number of spermatocytes were located at the edge of the follicular cavity, with a substantial number of spermatozoa arranged radially. These results confirm that the gonads of both sexes had reached the mature stage. Finally, on July 19, the female gonad (F(d)) showed pronounced yolk tissue staining, while the male gonad (M(d)) exhibited markedly diminished follicles, indicating that both gonads had reached the spawning. These observations confirm that both male and female A. pectinata reach their peak breeding period by late June.
-m(d)__male_gon.png)
3.2. RNA Sequencing and Assembly
Transcriptome sequencing of A. pectinata gonads yielded 3.1 GB and 2.4 GB of raw data for the female and male samples, respectively (Table 1). After rigorous quality control procedures, a total of 3.0 GB (female) and 2.25 GB (male) of high-quality clean reads were obtained. These reads were subsequently pooled for de novo assembly using Trinity, which generated 508728 transcripts with an N50 of 1036 bp and a mean length of 679 bp. Finally, 269916 coding sequences were predicted, and homology-based annotation revealed that 14.62% of the transcripts had significant matches to proteins from closely related species, such as Ostrea edulis and Crassostrea virginica, in the NCBI Nr database.
3.3. Genes Annotation
The 226,881 and 107,947 unigenes obtained were annotated in 7 databases, 53,651 unigenes (19.88%) were annotated in at least one database, and 1,674 unigenes were annotated in all databases (Table 2). With 14,261, 16,032, 12,131, 16,909, 16,932, 39,474, and 22,201 unigenes annotated in the CDD, PFAM, KEGG, KOG, GO, NR, and NT databases, respectively.
3.4. Analysis of Differentially Expressed Genes
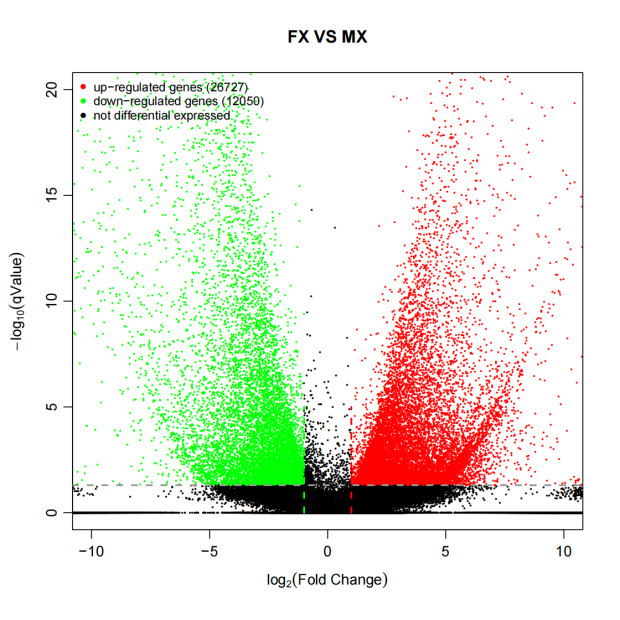
The DEGs were screened between the female and male gonadal transcriptomes. The results revealed that, compared to the male gonads, 26,727 genes were significantly up-regulated and 12,050 genes were significantly down-regulated in the female gonads (Fig. 2). Among these differentially expressed genes, a distinct subset exhibited a highly specific expression pattern. Specifically, 13,742 genes were identified as uniquely expressed in female gonads, while 1,851 genes were uniquely expressed in male gonads.
._red_and_green_dots_represent_upregulat.png)
3.5. GO and KEGG Enrichment Analysis
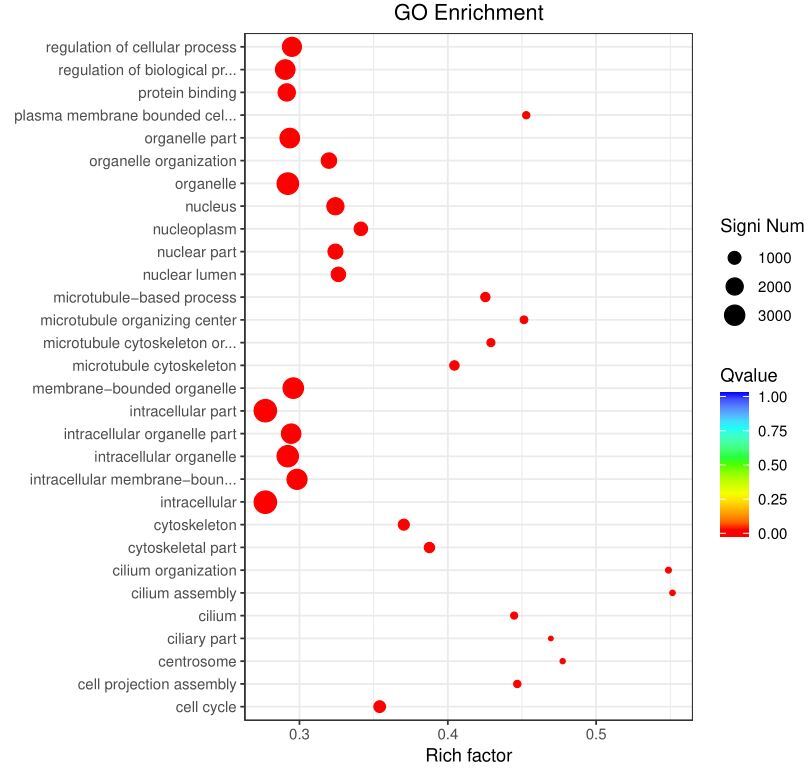
GO enrichment analysis revealed that DEGs were predominantly associated with cellular components (CC) such as organelles and intracellular components, biological processes (BP) including organelle organization, regulation of cellular processes, and regulation of biological processes, as well as molecular functions (MF) such as protein binding. Notably, approximately 50% of the genes enriched in cilium-related terms exhibited significant differential expression between male and female gonads (Fig. 3).
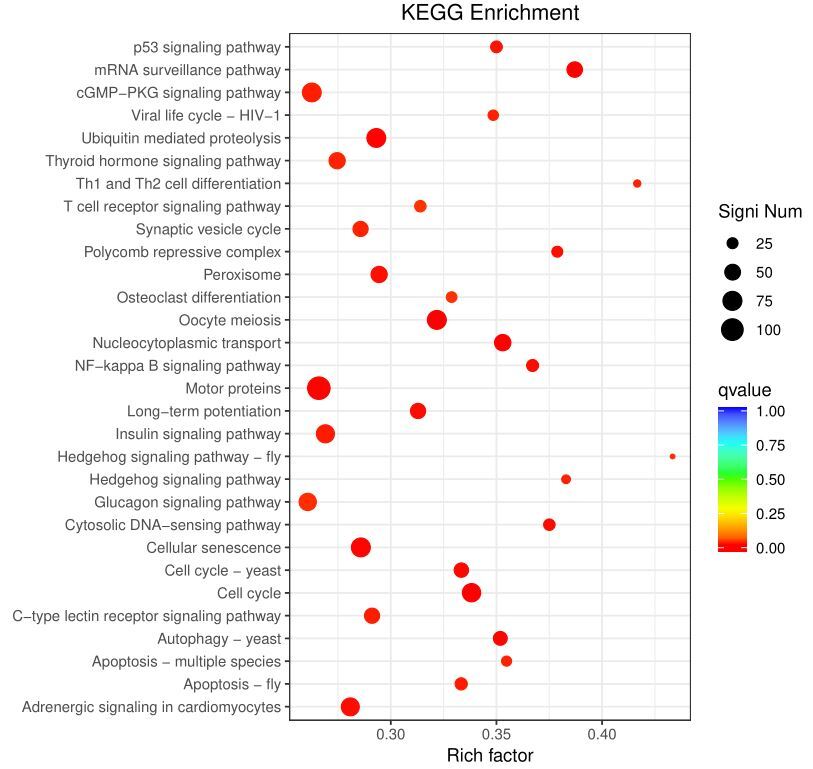
The DEGs were mapped to 302 KEGG pathways, of which 48 met the criteria for enrichment. Among the most enriched pathways, the cell cycle, cellular senescence, oocyte meiosis, ubiquitin-mediated proteolysis, motor proteins, adrenergic signaling in cardiomyocytes, insulin signaling pathway, cGMP-PKG signaling pathway, and glucagon signaling pathway stood out (Fig. 4).
_enrichment_analysis_for_differentially_expressed_genes_identified_bet.jpeg)
_pathway_enrichment_analysis_for_differentiall.jpeg)
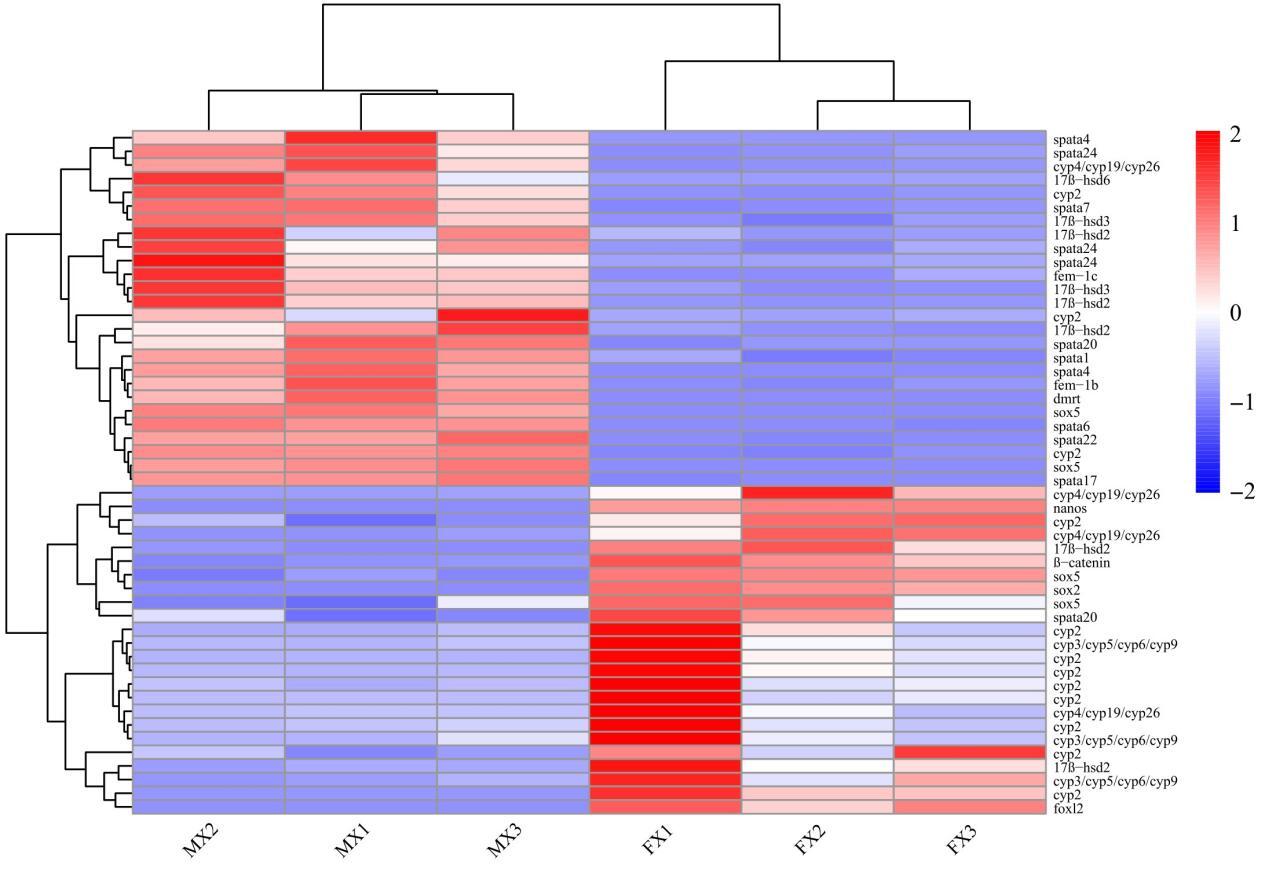
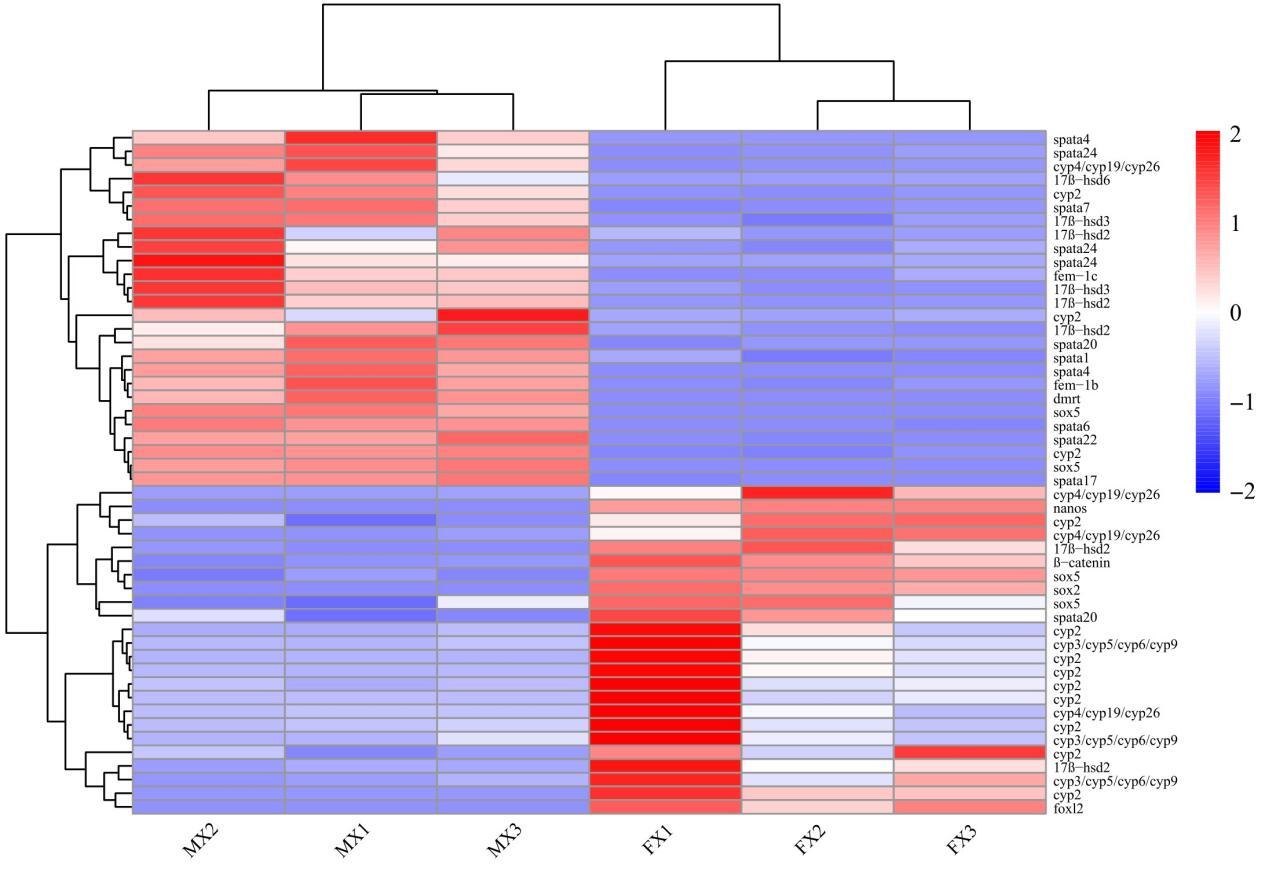
3.6. Key Sexual Differentially Expressed Genes
By screening differentially expressed genes (DEGs) between female and male gonads, we identified 50 regulatory factors closely related to sex determination and reproductive control (Fig. 5). Among them, Dmrt1, 17β-hsd (hsd6, hsd3), Spata (Spata1, Spata4, Spata6, Spata7, Spata17, Spata20, Spata22, Spata24), and Fem-1 (Fem-1b, Fem-1c) showed male-biased expression, whereas the Cytochrome P450 family (cyp2, cyp3/cyp5/cyp6/cyp9, cyp4/cyp19/cyp26), Foxl2, β-catenin, and Nanos exhibited female-biased expression. In the gene-expression clustering tree, Spata and Cyp/17β-hsd were located on the same branch, Dmrt and Sox5 on another branch, while Foxl2 was distant from the Dmrt branch.
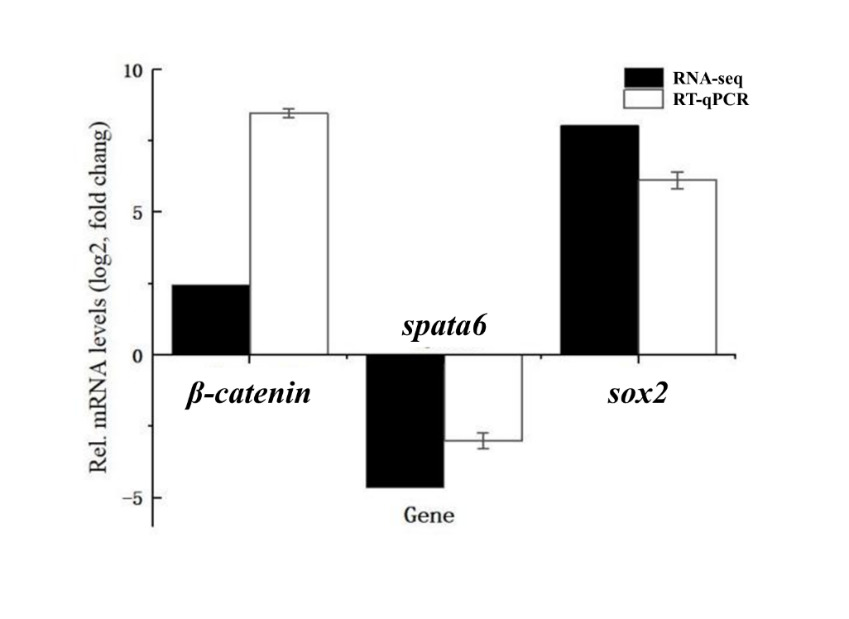
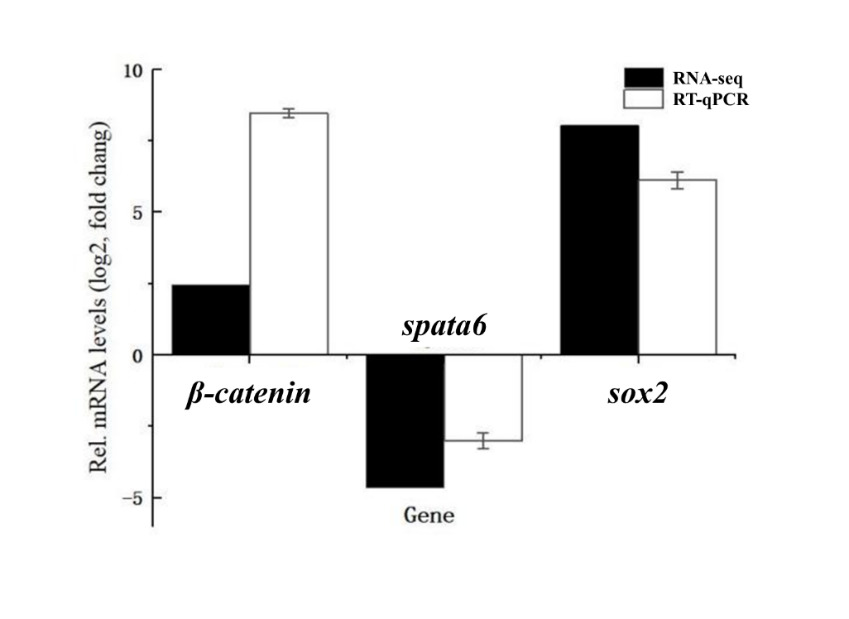
3.7. RT-qPCR Results
To validate the RNA-seq results, three DEGs (β-catenin, spata6, and sox2) were selected for RT-qPCR analysis. The RT-qPCR results showed that β-catenin and sox2 were upregulated in female gonads, while spata6 was downregulated compared to male gonads. These expression patterns were consistent with the transcriptome data, confirming the reliability of our RNA-seq analysis (Fig. 6).
4. Discussion
Atrina pectinata is an economically important bivalve mollusc; however, compared to other cultured species, such as oysters and scallops, its reproductive biology remains poorly understood, which constrains the development of large-scale artificial seed production. In the present study, histological sectioning revealed that the gonadal maturation peak of A. pectinata occurs in late June. Transcriptome sequencing was then employed to screen for sex-related differentially expressed genes (DEGs) and to dissect their underlying molecular regulatory mechanisms. Our results indicate that the regulatory network of A. pectinata is evolutionarily conserved yet exhibits complex molecular interactions, likely encompassing several functional modules including the DMRT1–FOXL2–β-catenin “bistable switch”, the DMRT1–FEM-1 ubiquitin-stabilization loop, the Nanos–β-catenin germ-cell–somatic-cell feedback loop, and the Spata–17β-HSD/CYP local-hormone–gametogenic coupling network.
The Dmrt gene family encodes conserved zinc-finger DM-domain transcription factors that are essential for sex determination and gonadal development.21 In the present study, Dmrt1 showed male-biased expression, consistent with findings in scallops,13,22,23 oysters,14,24 Hyriopsis cumingii25,26 and Haliotis asinina.27 Foxl2 possesses a highly conserved molecular structure28,29 and has been shown to regulate female gonadal development in Oplegnathus punctatus,30 Patinopecten yessoensis,31 Hyriopsis cumingii32 and Mulinia lateralis11; in the present study, Foxl2 exhibited significant female-biased expression in A. pectinata gonads. β-catenin, a key effector of the Wnt pathway, plays a crucial role in sex determination and gonadal differentiation, exhibiting female-specific expression in Crassostrea gigas33 and Hyriopsis cumingii.15 In the present study, β-catenin also showed significant female-biased expression. These conserved expression patterns suggest that Dmrt1, Foxl2 and β-catenin maintain evolutionarily conserved functions in A. pectinata.
The DMRT1–FOXL2–β-catenin “bistable switch” is considered the core circuit of vertebrate sex determination: Dmrt1 directly represses Foxl2, while Foxl2 binds to the Dmrt1 promoter and inhibits its expression, forming a positive feedback loop that locks in male or female fate; β-catenin occupies the Tcf/Lef sites of the Foxl2 promoter to enhance its transcription, further amplifying the female pathway and suppressing the DMRT1–SOX9 module.34,35 This circuit has been confirmed in molluscs: RNAi knockdown of Cg-Dmrt1 in Crassostrea gigas up-regulated Cg-Foxl2 by 4.3-fold and significantly enhanced β-catenin nuclear localization, providing the first direct evidence for a “Dmrt1-low → Foxl2-high → β-catenin nuclear signal strong” cascade in bivalves.17 In the present study, A. pectinata exhibited a similar expression pattern: high Dmrt1, low Foxl2 and low β-catenin in male gonads; low Dmrt1, high Foxl2 and high β-catenin in female gonads, implying that this circuit functions as an evolutionarily conserved “bistable switch” and serves as the core sex-determination module in A. pectinata.
In the classic nematode model, Fem-1 acts as the substrate-recognition subunit of the CUL-2–E3 ubiquitin complex to target TRA-1 (Dmrt ortholog) for degradation, thereby relieving repression of male programs.36 However, in the present study, Fem-1b and Fem-1c showed male-biased expression and co-expression with Dmrt1 in A. pectinata, opposite to the nematode paradigm. Similar findings have been reported in aquatic animals, including the male-biased expression of Fem-1b/c in Crassostrea gigas16 and the significant co-expression of Fem-1b/c with Dmrt1 in the male gonads of Hyriopsis cumingii.7 These observations suggest that the DMRT1–FEM-1 ubiquitin-stabilization loop may have evolved an “inverse” mechanism in bivalves, wherein Fem-1 no longer acts as a classical “degradation factor” but rather stabilizes Dmrt1 by repressing its ubiquitin-proteasome pathway, thereby reinforcing male fate determination and offering new perspectives for investigating sex plasticity in bivalves.
The RNA-binding domain of Nanos3 is highly conserved across grass carp, zebrafish, Atlantic salmon, mouse, and fruit fly.37 In Danio rerio, Nanos1/2/3 show female-biased expression and promote ovarian development and germ-cell formation38,39; in oysters, Nanos exhibits female-specific expression.40 In Crassostrea gigas, the 3′-UTR of Cg-Nanos carries two typical Tcf/Lef sites; overexpression of an activated β-catenin mutant increases nanos transcription by 2.1-fold, while Nanos protein binds Wnt4 mRNA to repress its translation, limiting somatic feminization signal diffusion and ensuring synchronous meiosis entry of germ cells and surrounding follicle cells17; similar findings have been reported in Sinonovacula constricta.41 In the present study, nanos were highly expressed in female gonads of A. pectinata and positively correlated with β-catenin, suggesting that the “β-catenin → Nanos → Wnt4 translational repression” feedback loop may also operate during female gonadogenesis in A. pectinata.
The Spata, 17β-Hsd, and CYP gene families exhibit high diversity across bivalve species and tissues.7,42,43 The Spata family has been detected in Gigantidas platifrons and Cristaria plicata, showing male-gonad-biased expression and being closely related to spermatogenesis, sperm motility and male sex maintenance.42 17β-HSD and CYP genes are essential for steroid hormone metabolism, synergistically maintaining sex-steroid hormone balance and regulating ovulation, gonadal development and sex determination.44–46 In bivalves, a metabolic–gametogenic coupling network involving the Spata family and 17β-Hsd/CYP has been reported: in Mactra lateralis, the male module shows co-expression of Spata1/4/6 with 17β-Hsd3, and in vitro incubation confirmed that the 17β-HSD3-catalysed androstenedione → testosterone step increases Spata6 promoter activity by 1.8-fold, indicating that local androgen production is sufficient to activate the spermatogenesis programme directly47; in Ruditapes philippinarum, the female module involves CYP19A1 (aromatase) converting testosterone to 17β-estradiol, co-modules with Foxl2/β-catenin and simultaneously represses Dmrt1 and Spata, achieving a “metabolic–sex” dual feminization.48 In the present study, CYP and Spata were negatively correlated in female gonads of A. pectinata, whereas 17β-Hsd and Spata were positively correlated in males, consistent with the expression patterns reported above, implying that a Spata–17β-HSD/CYP metabolic–gametogenic coupling network may also operate in A. pectinata.
Sox gene family members play important roles in gonadal development across fish, shrimp, crab and molluscs.49 Studies on fish and bivalves have revealed the existence of a SOX–DMRT co-regulation axis: in zebrafish, Sox5 directly represses Dmrt1 transcription to regulate testis–ovary switching50; in Crassostrea gigas and Patinopecten yessoensis, SoxH and Dmrt1 are both significantly higher in mature male gonads than in females.51,52 In the present study, we identified five sox2 and sox5 homologues in A. pectinata, but neither showed consistent sex-biased expression, possibly due to the functional diversity of the Sox gene family.53–55 Nevertheless, the significant gonad-biased expression levels of most Sox2/5 homologues still imply a strong association between the Sox gene family and sex in A. pectinata; whether a SOX–DMRT co-regulation axis exists awaits further functional validation.
In the present study, we performed transcriptome profiling of the peak gonadal development stage of A. pectinata, constructed the corresponding gene-expression landscape, and screened a series of sex-related differentially expressed genes (DEGs). By analyzing potential sex–related and reproduction-related molecular regulatory mechanisms, we found that the molecular regulatory network of A. pectinata is evolutionarily conserved yet exhibits species-specific characteristics. Our results provide preliminary insights into the molecular mechanisms of sex determination and gonadal development in A. pectinata, offer candidate genes and key regulatory modules for in-depth mechanistic studies, and serve as a reference for reproductive biology and genetic breeding of this species. Owing to the preliminary screening nature of this study, only a single developmental time-point was examined;consequently, our conclusions cannot reflect dynamic gene-expression patterns, and direct molecular interactions among genes still require functional validation. In the next step, we will focus on the dynamic expression of key regulatory factors and verify their mutual interactions.
Acknowledgments
This work was financially supported by the Yantai Marine Economic Research Institute, Yantai, PR China, and School of Fisheries, Ludong University, Yantai, PR China. This research was funded by the Yantai Science and Technology Innovation Development Plan (Project No. 2023JCYJ100); the National Natural Science Foundation of China (Grant No. 42076088 & 42406127); Key Research and Development Program of Shandong Province (No. 2022LZGC015); Special Funds for the Taishan Scholar Project of Shandong Province, China (No. tstp20240518); Special Supporting Funding for Leading Talents above the provincial level in Yantai City (No. 202203); and Modern Agricultural Industry Technology System of Shandong Province, China (No. SDAIT-14-01).
Authors’ Contribution
Methodology: Cong Wei (Lead). Formal Analysis: Cong Wei (Equal), Lufei Yang (Equal), Deyan Hu (Equal). Investigation: Cong Wei (Equal), Yan Gao (Equal), Lufei Yang (Equal), Deyan Hu (Equal), Hongce Song (Equal), Junsong Yangli (Equal), Yijing Han (Equal). Writing – original draft: Cong Wei (Equal), Yan Gao (Equal). Conceptualization: Yan Gao (Equal), Xiaotong Wang (Equal), Wei Chen (Equal). Visualization: Lufei Yang (Equal), Deyan Hu (Equal). Writing – review & editing: Xiaotong Wang (Equal), Wei Chen (Equal). Supervision: Xiaotong Wang (Equal), Wei Chen (Equal). Funding acquisition: Xiaotong Wang (Equal), Wei Chen (Equal).
Competing Interest
The authors declare no conflicts of interest.
Ethical Conduct Approval – IACUC
Ethical review and approval were waived for this study due to A. pectinata belonging to invertebrates.
Informed Consent Statement
All authors and institutions have confirmed this manuscript for publication.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding authors.