


1. Introduction
Sea urchin (Strongylocentrotus intermedius) is a characteristic cold-water species whose optimal growth temperature ranges from 15 ℃ to 20 ℃. Previous studies have shown that temperature is a key factor influencing sea urchin physiology.1,2 Prolonged exposure to temperatures above 23 ℃ induces severe physiological stress in S. intermedius, manifesting as reduced feeding, suppressed growth, and increased mortality.3–5
The gut bacterial communities influence host nutrient metabolism, immune responses, and growth performance, whereas their composition and structural integrity are susceptible to environmental temperature fluctuations.6–8 The temperature fluctuations often induce gut dysbiosis in aquatic species, ultimately compromising host health.9,10 For instance, in the Yesso scallop (Patinopecten yessoensis), exposure to elevated temperature induces intestinal tissue damage along with gut microbial dysbiosis, characterized by an increased abundance of potential pathogens such as Tenacibaculum and MycoplasmaIn.11 In red swamp crayfish (Procambarus clarkii), high temperatures affect gut microbiota composition, including an increase in pathogens like Citrobacter, and regulate the levels of intestinal metabolites, such as sugars and amino acids.12 In rainbow trout (Oncorhynchus mykiss) revealed that elevated temperatures (18-21 ℃) reduced relative abundances of Proteobacteria and increased Firmicutes, impairing metabolism and gut barrier function.13,14 In sea cucumbers (Apostichopus japonicus), exposure to 26 ℃ significantly increased the relative abundance of Firmicutes and Desulfobacterota in the gut microbiota, which may disrupt ecological balance and impair host health.15 Therefore, it is essential to understand the effect of temperature on the gut bacteria community of S. intermedius. Also, the gut microbiota exerts its influence on the host through complex, interactive networks among bacterial species rather than monospecific colonies. Therefore, when environmental fluctuations perturb host physiology, the preservation of microbial community function becomes more contingent upon the structural stability of the microbial community network.16 Analysis of its network characteristics thus facilitates the assessment of community resistance to disturbance and the elucidation of underlying functional response mechanisms.
Network analysis serves as a pivotal tool for analysing microbial interactions and assessing community stability.17–19 Quantifying key topological metrics, such as complexity, modularity, and core taxa, enables the evaluation of community ecological stability, the prediction of ecological function, and the elucidation of stress response mechanisms.20 Previous studies have shown that exposure to sulfamethoxazole for 30 days significantly reduced the complexity of the gut bacterial network in marine medaka (Oryzias melastigma), with the number of nodes, edges, and modularity decreasing from 76, 308, and 0.593 to 53, 198, and 0.505, respectively.21 Under temperature fluctuations, the gut bacterial network in the mud crab (Scylla paramamosain) exhibited increased complexity, with the average path length rising from 2.42 to 3.29 and the clustering coefficient from 0.73 to 0.75, indicating a restructured pattern of microbial interactions.22 Furthermore, in sea cucumbers (A. japonicus), the probiotic Paracoccus marcusii DB11 promoted the integration of sub-modules within the gut bacterial network, increasing interspecies interactions and the number of connectors and module hubs. In contrast, the antibiotic florfenicol reduced connector numbers and interspecies interactions, thereby disrupting network integrity.23 Network analysis is a key method for revealing the mechanisms by which bacterial communities respond to environmental stress and for understanding the ecological functions of microbial communities and their interactions with the host.
Although the influence of temperature on the microbiota of aquatic animals is well recognized, the characteristics of the gut bacterial community in sea urchins under temperature remain unclear. Therefore, this study investigated the structural and co-occurrence network characteristics of the gut bacterial community in S. intermedius under different temperatures (13, 16, 19, 22, and 25 ℃). These findings provide novel insights into the ecological adaptations of the gut bacterial community in sea urchins facing temperature fluctuations.
2. Materials and methods
2.1. Experimental design
A total of 600 healthy 6-month-old sea urchins (S. intermedius) with a diameter of 3.00 ± 0.20 cm were acclimated for two weeks in a recirculating aquaculture system (RAS). The conditions during acclimation were maintained at temperature (19.0 ± 1.0 ℃), salinity (30.0 ± 1.0), dissolved oxygen (6.9 ± 0.2 mg/L), and pH (8.0 ± 0.1). Following acclimation, individuals were randomly assigned to five temperature treatment groups (13℃, 16℃, 19℃, 22℃, and 25℃). Three parallel sets were set for each group, with 40 sea urchins per tank (n=40 per replicate). Throughout the experimental period, seawater was sand-filtered. The sea urchins were fed fresh Ulva lactuca. Feces were removed by siphoning every two days, and only one-fifth of the water was renewed to minimize disturbance and maintain a stable thermal environment. The water temperature in each treatment group was adjusted from the acclimation temperature to its respective target level at a rate of 1.0 ℃ per day and was thereafter maintained with fluctuations within ± 0.2 ℃.
2.2. Sample collection and DNA extraction
After the end of the 30-day experiment, a total of 15 sea urchins were randomly selected from each temperature group (5 individuals per replicate) for dissection. Prior to dissection, each sea urchin was rinsed with sterile seawater.24 The gut tissues were then removed in a biosafety cabinet, wiped with 75% alcohol on the surface, and placed into 1.5 ml sterile centrifuge tubes. To minimize individual variation, the intestinal tissues from five sea urchins in each replicate were pooled to form a single composite sample. Then, frozen in liquid nitrogen and stored at -80℃. All the instruments used in the experiment were autoclaved to sterilize them. The gut samples from the 13 ℃, 16 ℃, 19 ℃, 22 ℃, and 25 ℃ groups were labeled as G13, G16, G19, G22, and G25, respectively.
The gut samples were minced using sterile scissors and transferred to a 2 mL centrifuge tube. To each tube, an appropriate amount of steel beads and 800 μL of SLX buffer were added. The samples were then homogenized using a tissue grinder. DNA extraction was subsequently performed following the protocol provided with the E.Z.N.A.™ Mag-Bind Soil DNA Kit (OMEGA). Negative controls were added during the DNA extraction to prevent contamination.
2.3. 16S rRNA gene sequencing
The extracted DNA was quantified using a NanoDrop ND-2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA) and analyzed by electrophoresis on 1.0% agarose gels. The V3-V4 region of the bacterial 16S rRNA gene was amplified using the primer pairs 341F (5’-CCTACGGGNGGCWGCAG-3’) and 805R (5’-GACTACHVGGGTATCTAATCC-3’). The PCR amplification protocol followed that described in a previous study.25 The PCR products were resolved on 2.0% agarose gels and purified using Agencourt AMPure XP Beads (A63881, Beckman, USA). The DNA samples were then submitted for paired-end Illumina sequencing.
2.4. Sequence analysis
Bioinformatics analysis of microbial communities was conducted using QIIME2 (v2019.4). Raw amplicon sequences were subjected to quality filtering and adapter trimming. Amplicon Sequence Variants (ASVs) were inferred through the DADA2 pipeline for sequence denoising. Taxonomic assignment of the 16S rRNA gene ASVs was performed against the Greengenes database (version 13.8) using the naïve Bayesian classifier implemented in DADA2.26 Alpha diversity indices (Chao1, Observed_species, Shannon, Simpson) and beta diversity (based on Bray-Curtis distance) were calculated, with statistical testing performed via PREMANOVA. Putative gene functions were predicted using PICRUSt (v1.0.0) with KEGG databases.
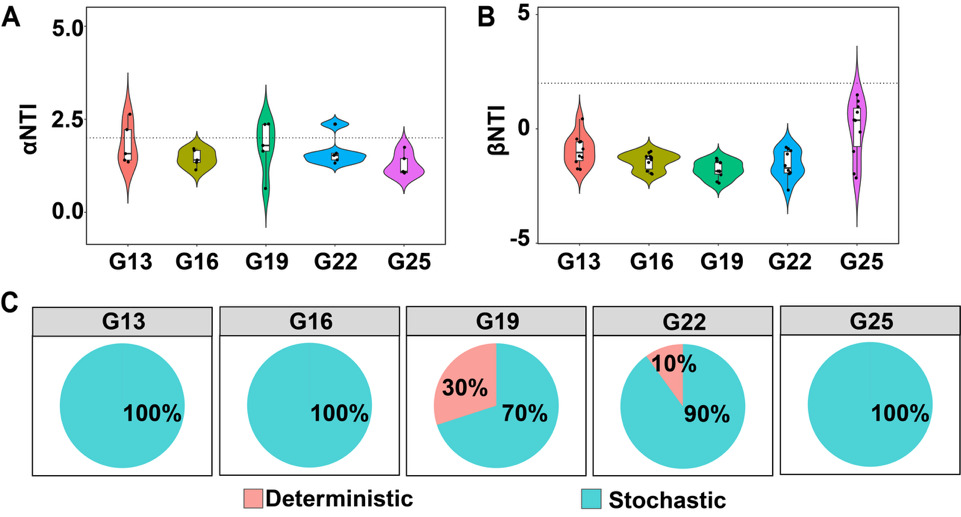
Co-occurrence networks were constructed using Spearman rank correlations ( | r | > 0.60, P < 0.05), visualized with Gephi, and analyzed for key network topology metrics. Community assembly processes were assessed by calculating the Nearest Taxon Index (βNTI) via the picante package27; | βNTI | > 2.00 indicates deterministic dominance, whereas | βNTI | < 2.00 reflects stochastic dominance. All statistical analyses and visualizations were performed in R.
2.5. Statistical analysis
All data are presented as the mean ± standard deviation (AVG ± SD). All data were tested for normality using the Kolmogorov-Smirnov test for normality and Levene’s test for homogeneity of variances. One-way analysis of variance (ANOVA) and the LSD test were performed using SPSS 26.0.
3. Results
3.1. Sequencing results
In this study, sequencing of the sample targeting the 16S rDNA gene yielded an average of 80,828 raw sequences. After quality filtering, an average of 73,975 sequences were obtained. The proportion of valid sequences exceeded 96.94% with a coverage rate of 99.13%, confirming that the sequencing results accurately reflected the biological characteristics of the samples.
3.2. Diversity analysis of gut bacteria communities of sea urchins
A Venn diagram was used to illustrate the unique OTUs in the gut samples (Figure 1B). Specifically, 998, 387, 275, 683, and 1,671 representative OTUs were identified in the G13, G16, G19, G22, and G25 groups, respectively. In total, 9 OTUs were shared among all five groups, while 532, 224, 135, 365, and 910 OTUs were unique to the G13, G16, G19, G22, and G25 groups.
Chao1 index, Observed_species index, Simpson index, and Shannon index were used to evaluate the alpha diversity and richness of gut bacteria communities (Figure 1A). Compared with the other groups, G13 and G25 exhibited significantly greater diversity and richness of the gut bacterial community. In contrast, the G19 group showed the lowest species diversity and richness (P < 0.05). PCoA based on Bray–Curtis distances revealed that the PCoA1 and PCoA2 accounted for 52.20% and 20.90% of the total variation, respectively (Figure 1C). Significant differences in sea urchin gut bacterial communities were observed with changes in the water temperature (P < 0.05).

3.3. Structural characteristics of the gut bacteria community
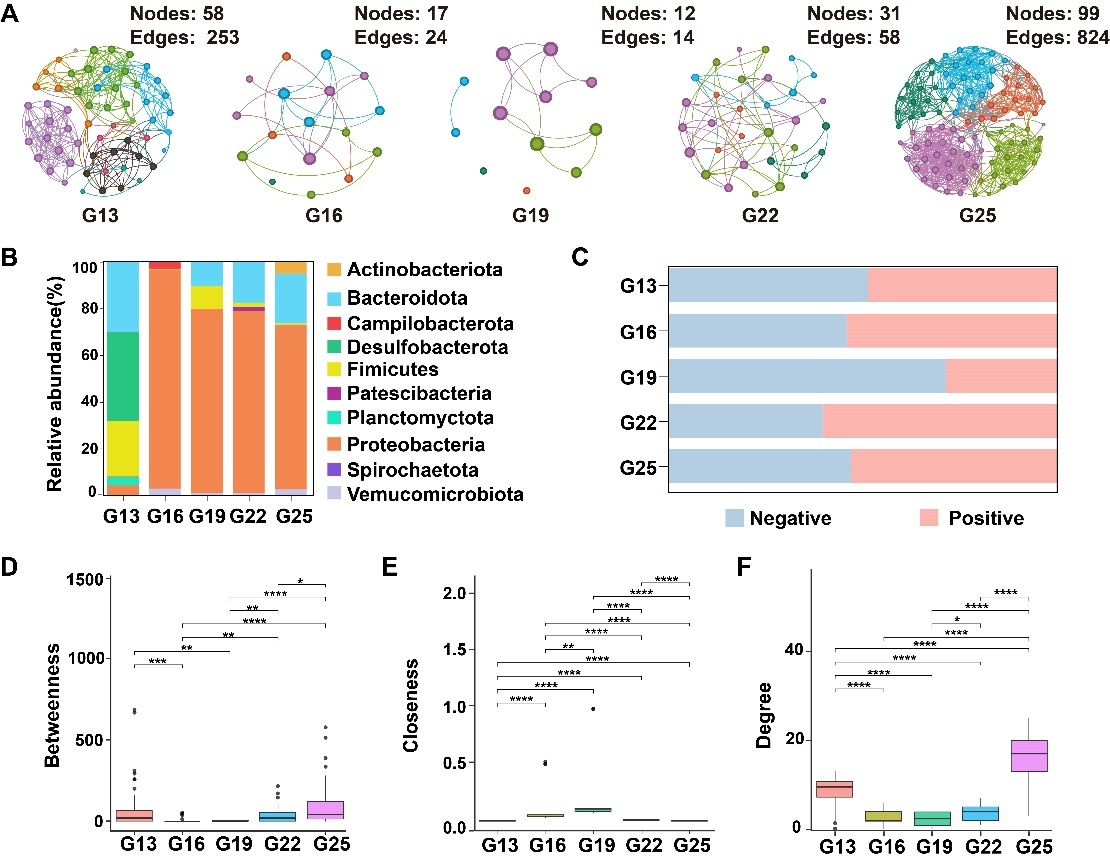
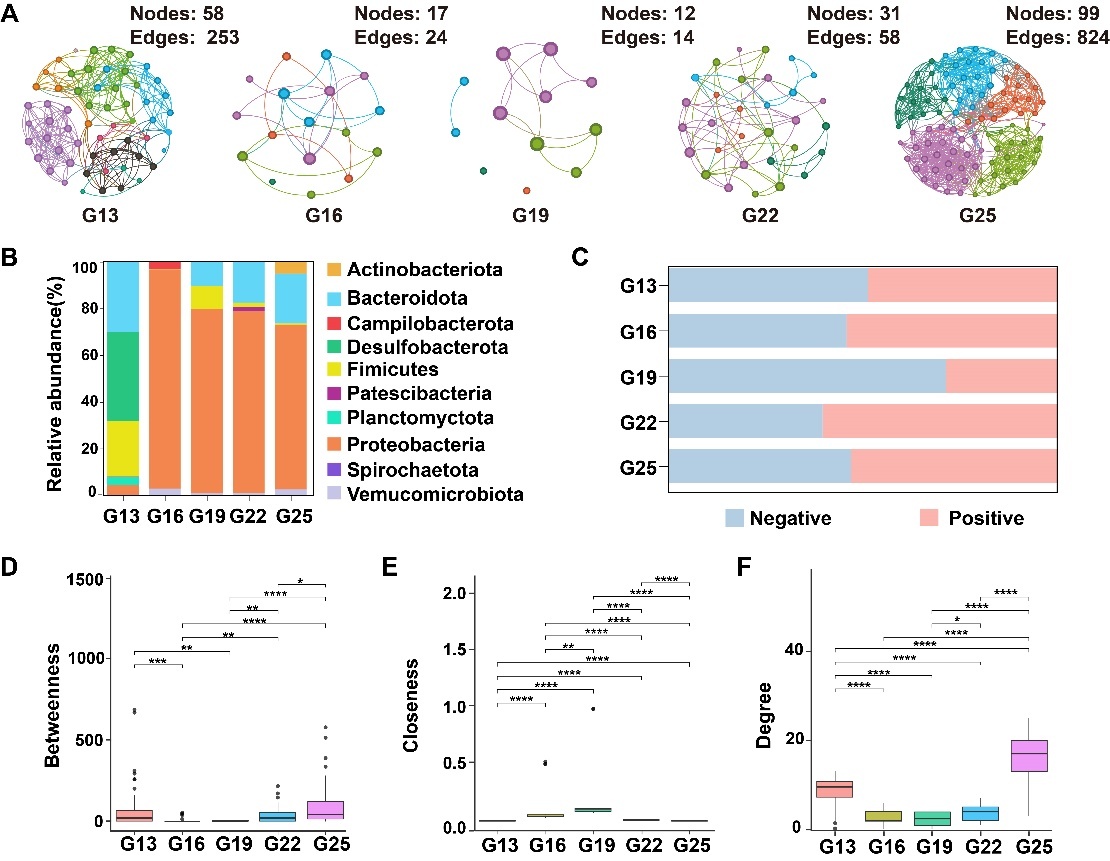
This study demonstrated significant differences in the gut bacterial community of S. intermedius across different temperatures. At the phyla, the highest relative abundance of gut bacteria communities were Proteobacteria, Bacteroidota, and Firmicutes (Figure 1D). The relative abundance was highest for Firmicutes in the G13 group (22.76%), while Proteobacteria was highest in the G19 group (95.14%), and Bacteroidota was highest in the G25 group (21.98%). At the genus level, Ruegeria, Desulforhopalu, and Pseudomonas were the dominant genera in the gut bacteria community of the sea urchin. The relative abundance of Pseudomonas was highest in the G19 group (31.49%) and lowest in the G13 group (9.56%) (Figure 4). Desulforhopalus was most abundant at G13 group (34.25%), while it was almost not detected in the other groups. Cohaesibacter was found to have higher relative abundances in the G16 (11.71%). The relative abundance of Ruegeria was highest in the G22 and G25 groups (30.21% and 33.23%, respectively), and it was also detected in the G16 group (Figure 1E).
The LEfSe analysis shows that the gut bacterial communities selectively enrich bacteria with specific capabilities to adapt to different temperature environments (Figure 2). In the G13 group, Desulfobacteria were significantly enriched, including the genus Desulforhopalus. In the G16 group, Variovorax and Paracoccus were enriched; both have versatile metabolic capabilities. In the G19 group, Proteobacteria were enriched, specifically the family Pseudomonadaceae, and the genus Pseudomonas. At the G22 group, Barnesiella and Rhodopirellula were enriched. Notably, Pseudophaeobacter, Ruegeria, Sphingomonas, and Burkholderia_Caballeronia_Paraburkholderia were significantly enriched in the G25 group.

3.4. Functional prediction of the gut bacterial communities
Based on the KEGG database, a total of 170 tertiary pathways with significant differences (P < 0.05) were obtained in the five groups, which could be classified into 33 secondary metabolic pathways and 6 primary metabolic pathways. Differential metabolic pathways were carbohydrate metabolism, amino acid metabolism, cofactor and vitamin metabolism, and DNA replication and repair. The G19 group exhibited significant upregulation in several pathways, including the digestive system, lipid metabolism, glycan biosynthesis and metabolism, terpenoid and polyketide metabolism, folding, sorting, and degradation, and bacterial infections (P < 0.05). The G25 group showed significant enrichment in signaling molecules and interactions compared to the other groups. The G13 group’s immune system pathways showed significant differences compared with those of the other groups (Figure 3).
_at_different_temperatures.svg)
3.5. Co-occurrence networks of different temperature groups
Based on the structural topology analysis of the sea urchin gut bacterial communities, it was concluded that the symbiotic network of gut bacteria in the G25 group was significantly larger than those in the other groups (Figure 4). Specifically, the nodes in the G13, G16, G19, G22, and G25 groups were 58, 17, 12, 31, and 99, respectively, while the edges were 253, 24, 14, 58, and 824 (Table 1). The G13 and G25 groups exhibited higher network complexity, characterized by increased nodes and edges as well as higher average degree (avgK) values. The modularity of the G13 group network was the highest (0.616) and contained 8 modules. In comparison, the G16, G19, G22, and G25 groups each contained 5 modules, with modularity coefficients of 0.556, 0.309, 0.592, and 0.609, respectively (Figure 4A). Degree centrality and betweenness centrality increased from the G16 to the G25 group, whereas closeness centrality was significantly higher in the G19 group than in the other temperature groups (Figure 4D-F). The co-occurrence networks of the G13, G19, and G25 groups showed balanced proportions of positive and negative microbial interactions (Figure 4C). The G19 and G25 groups had higher percentages of positive interactions (54.65% and 53.71%, respectively), while the G22 group was dominated by positive interactions (60.28%). In contrast, negatives were most prevalent in the G16 group (71.92%). A symbiotic network was established, with Proteobacteria accounting for 67.10%, Bacteroidetes for 14.71%, and Firmicutes for 7.95%. (Figure 4B).
In our study, the α-NTI values were positively correlated with the temperature fluctuation, indicating phylogenetic clustering of bacteria with symbiotic relationships in the bacterial community of sea urchin was greater than expected, with a more pronounced effect observed at the G19 group (Figure 5A). The β-NTI (phylogenetic turnover rate) was significantly higher in G19 group gut samples than in other groups, indicating that the gut bacterial community assembly of S. intermedius was predominantly shaped by stochastic processes (Figure 5B). Notably, deterministic processes mainly drove the assembly of the gut bacterial community in the G19 group (Figure 5C), indicating an increased selective effect of temperature on the gut bacterial community of the G19 group.
_at_differen.png)
4. Discussion
4.1. Effects of different temperatures on the gut bacteria community of sea urchin
The gut bacterial community plays a crucial role in mediating host adaptation to environmental changes, with temperature being a key factor influencing microbial composition and function.28 In this study, the gut bacteria of S. intermedius exhibited clear temperature-dependent structural shifts at both phylum and genus levels, reflecting adaptive microbial restructuring in response to thermal variation.
At the phylum level, Proteobacteria, Bacteroidota, and Firmicutes constituted the core phyla and consistently dominated the gut microbiota across all temperature groups. This aligns with previous reports identifying these taxa as major components of the bacterial community in S. intermedius. Consistent with our findings in S. intermedius, studies indicate that these bacteria represent the predominant component of the bacterial community in the benthic environment by S. intermedius.29 The relative abundance of these phyla, however, varied significantly with temperature. Under the optimal growth temperature of 19 °C (G19), the relative abundance of Proteobacteria reached in 95.14%. Previous studies have reported that the feeding rate of S. intermedius is highest at this temperature.30 As one of the largest and metabolically most versatile prokaryotic groups,31 the high metabolic plasticity of Proteobacteria likely plays a key role during periods of active host feeding: by enhancing the degradation of complex organic matter, it may directly support improved host nutrient metabolism and energy acquisition.32,33
In contrast, the relative abundance of Firmicutes in the G13 group increased significantly to 22.76%, suggesting a metabolic adaptation to low-temperature stress. Firmicutes are known to efficiently ferment carbohydrates to produce utilizable energy sources such as short-chain fatty acids.34 Under low-temperature conditions that reduce host metabolic rate and constrain energy intake, the rise in Firmicutes abundance could help sustain host energy supply by enhancing intestinal fermentation, thereby alleviating physiological pressure induced by cold stress.35
The relative abundance of Bacteroidota reached 21.98% in the G25 group, a change that may be linked to the host’s physiological adaptation under high-temperature stress. Bacteroidota are enriched with polysaccharide-degrading enzymes, enabling efficient breakdown of dietary fiber and complex carbohydrates into metabolites such as short-chain fatty acids (SCFAs), which can serve as an energy source for the host.36,37 Under high temperatures, host metabolic rate typically increases, accelerating energy expenditure, while feeding efficiency may decline, potentially leading to an imbalance between energy supply and demand.38 The observed increase in Bacteroidota abundance may therefore enhance intestinal polysaccharide degradation, improving energy harvest efficiency and helping to mitigate energy metabolic pressure induced by heat stress, thereby contributing to the maintenance of gut homeostasis. Furthermore, certain members of Bacteroidota have the potential to modulate host immune and inflammatory responses,39 suggesting that changes in their abundance may also contribute to the host’s overall physiological regulation during thermal challenge.
Genus-level analysis further elucidated distinct temperature-dependent enrichment patterns associated with functional adaptation. In the G13 group, the psychrophilic sulfate-reducing genus Desulforhopalus dominated (34.25%). Its known capacity for gas vacuole formation and organohalide respiration suggests that it may contribute to energy provision in cold environments, potentially through pathways such as 2,6-dibromophenol debromination, thereby supporting host metabolic maintenance during low-temperature periods.40,41 At the optimal temperature (G19), Pseudomonas became the dominant genus (31.49%). Although traditionally regarded as an opportunistic pathogen,42,43 its sustained dominance in healthy individuals implies a potential dual role as both a conditional pathogen and a beneficial commensal. This functional plasticity aligns with reported oxygen-dependent bacteriostatic activity of Pseudomonas against fermentative bacteria, highlighting the context-dependent nature of its ecological role in marine invertebrates.44 It has been demonstrated that Ruegeria and Sphingomonas play a protective role against environmentally induced microbial dysbiosis in diverse marine animals. Prior studies indicate that these bacterial genera contribute to microbiome stability, particularly in marine hosts, by producing quorum-quenching enzymes and antimicrobial compounds. Specifically, Ruegeria produces N-acylhomoserine lactonases that quench pathogen quorum-sensing signals and suppress virulence expression, and further contribute to host microbial homeostasis by modulating the associated bacterial community to counteract pathogenic proliferation while promoting probiotic taxa, thereby alleviating Vibrio-induced dysbiosis.45,46 Similarly, Sphingomonas have been reported to harbor quorum-quenching enzyme genes and to produce antimicrobial substances, demonstrating antagonistic activity against Vibrio anguillarum in fish, thereby improving host survival and immune parameters.47 In this study, the enrichment of Ruegeria and Sphingomonas in the sea urchin gut under high temperature may indicate the activation of a similar microbiota-assisted defense mechanism, thereby contributing to the maintenance of intestinal microbial community stability. However, the specific functions of these bacteria in our study and their direct contribution to the sea urchin remain to be further investigated.
4.2. Effects of different temperatures on the gut symbiotic bacteria community of sea urchins
Microbial co-occurrence network analysis provides a methodological framework for linking ecosystem complexity to stability, offering insights into community structure and species interactions.48,49 Community stability is often associated with topological features such as a high average clustering coefficient and increased modularity.50,51 In this study, temperature fluctuations altered network structure, as evidenced by changes in edge numbers and modularity. Higher modularity in the G13 and G25 groups indicates the formation of densely connected, functionally coherent modules, which may enhance community stability and resilience under external stress. In contrast, the G19 group exhibited the lowest modularity, with a network dominated by negative correlations, reflecting competitive interactions. This structural simplification likely results from temperature acting as an environmental filter, excluding less tolerant taxa and reducing niche overlap and functional redundancy.52
The average path length (APL) reflects network connectivity and the efficiency of microbial interaction. An increased APL in the G13 group suggests longer interaction pathways, which may reduce metabolic efficiency and delay coordinated community responses to environmental stress such as low temperature.53,54 Such functional impairment could hinder the host’s nutrient acquisition and metabolic homeostasis.55 Ecological networks comprise both positive (e.g., mutualistic) and negative (e.g., competitive) interactions.56 In this study, the G16, G22, and G25 groups showed a higher proportion of positive correlations, suggesting enhanced cooperative exchanges. Conversely, the G13 and G19 groups showed stronger negative correlations, indicating competitive dominance. Although competition can contribute to network robustness,57 higher modularity and a balance of interaction types likely support more stable coexistence.58 Thus, the intestinal microbial community appears most stable in the G13 and G25 groups, where temperature-induced shifts promoted either competitive structuring or cooperative coherence, each favoring network persistence.
Community assembly analysis revealed that stochastic processes predominantly drove microbiota clustering across temperatures, except in the G19 group, where deterministic selection was observed. Under suboptimal temperatures, increased stochasticity may arise from a balance between microbial loss and gain,59 possibly as cooperatively adaptive strategies emerge.
Collectively, these findings indicate that temperature critically shapes the assembly and stability of the sea urchin intestinal microbiota, with the G13 and G25 groups exhibiting more stable network architectures. These observed changes warrant further exploration in future studies.
4.3. Temperature-dependent microbial strategies and host adaptation
The alpha diversity of the bacterial community was lowest at G16 and G19 groups, which may, conversely, indicate a stable state of the intestinal community. Previous studies have shown that the temperature range of 16-19 ℃ is the optimal range for sea urchin growth and feeding.60 Within this optimal temperature range, enhanced feeding activity and stable host metabolism may selectively promote the enrichment of specific core microbial taxa, thereby reducing overall diversity.61 In this study, the significant enrichment of Pseudomonas at G16 and G19 groups may be due to its unique metabolic functions. Research has shown that Pseudomonas can decompose complex nutrients and assist the host in energy acquisition, thereby promoting growth performance, enhancing immune function, and optimizing gut health, thus enhancing the host’s health and disease resistance.62,63 Their dominance under optimal temperatures may therefore facilitate more efficient nutrient absorption and contribute to the host’s growth and feeding performance.
In contrast, the higher diversity and more complex network structures observed in the G13, G22, and G25 groups are likely driven by distinct mechanisms. In the G13, G22, and G25 groups, the decline in host physiological integrity and its selective pressure on the bacterial community, coupled with potential colonization by opportunistic or stress-tolerant generalist bacteria, could lead to increased species diversity and more complex network structures.64 In the G13 group, the significant enrichment of Desulforhopalus may represent a microbial adaptation to low-temperature physiological stress. Desulforhopalus is an anaerobic bacterium specializing in sulfate reduction. This metabolic process provides the bacterial community with essential energy when the host’s metabolism slows at low temperatures.65,66 Furthermore, it helps maintain an anaerobic gut environment, which may help suppress colonization by harmful aerobic pathogens.67,68 Under high-temperature conditions (G25), the significant enrichment of potential beneficial bacteria (Ruegeria and Sphingomonas) within the gut bacterial community of S. intermedius may represent a compensatory microbial response. While the extent of their functions remains to be further investigated, these compositional adjustments suggest a potential functional reorganization of the microbiome that could help mitigate thermal stress.69,70 This provides crucial microbial support for the host to adapt to high-temperature stress.
5. Conclusion
In conclusion, the gut bacteria community of sea urchins exhibits distinct temperature-dependent characteristics, with Proteobacteria, Bacteroidetes, and Firmicutes constituting the core taxa in the symbiotic network. Changes in the gut bacterial community are closely linked to the complexity of the gut symbiotic network in the sea urchin. This study, through a 30-day experiment, has provided a foundational understanding of the sea urchin bacterial community’s response to temperature changes. However, due to significant seasonal and long-term temperature fluctuations in natural aquaculture environments, further long-term studies are needed to better understand the influence of the bacterial community on the host’s temperature adaptability.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2024YFD2401803), The Major Agriculture Project of Science and Technology Department of Liaoning Province (2023JH1/10200007), The National Natural Science Foundation of China (32373109), and The National Key Research and Development Program of Dalian (2022YF16SN067).
Authors’ Contribution– Credit Taxonomy
Methodology: Tengyu Xing (Lead). Formal Analysis: Tengyu Xing (Equal), Luo Wang (Equal), Dongsheng Chen (Equal). Investigation: Tengyu Xing (Equal), Dongsheng Chen (Equal), Yuchen Chen (Equal). Writing – original draft: Tengyu Xing (Lead). Writing – review & editing: Tengyu Xing (Lead), Anzheng Liu (Equal). Conceptualization: Luo Wang (Lead). Funding acquisition: Luo Wang (Lead). Supervision: Luo Wang (Lead). Resources: Xinyao Chen (Equal), Hai Zhang (Equal), Mengqi Li (Equal).
Competing of Interest – COPE
No competing interests were disclosed.
Ethical Conduct Approval – IACUC
The Institutional Animal Care and Use Committee (IACUC) of Dalian Ocean University does not require authorization for the research on Strongylocentrotus intermedius and other echinoderms. During the farming and dissections, measures were taken to minimize animal suffering, such as providing water conditions that mimic the natural environment and performing rapid dissections.
Informed Consent Statement
All authors and institutions have confirmed this manuscript for publication.
Data Availability Statement
All are available upon reasonable request.