
Introduction
Marine annelids are important components of benthic ecosystems and contribute substantially to sediment bioturbation, nutrient cycling, and community dynamics across coastal to deep-sea habitats.1,2 Within the order Eunicida, the family Onuphidae represents a diverse and ecologically significant group of tube-dwelling polychaetes that inhabit soft sediments and play pivotal roles in benthic structuring and energy flow.3 Members of the subfamily Hyalinoeciinae, including the genus Hyalinoecia, are distinctive for their free-living epibenthic lifestyle; unlike most Onuphinae, they are mobile epibenthic species that construct quill-like tubes from which they protrude and move in a caterpillar-like fashion known as epibenthic crawling, thereby facilitating mobility on the seafloor.2,4 Hyalinoecia robusta Southward, 1977, a deep-sea quill worm often found at depths of 400–1,600 m in muddy sediments, exhibits unique life-history traits such as simultaneous hermaphroditism with an adolescent male phase and annual iteroparous reproduction.2,5 Despite its ecological relevance in deep-sea benthic communities, genomic resources for H. robusta and many other onuphid taxa remain scarce, limiting insights into their evolutionary history and functional adaptations.
Animal mitochondrial genomes (mitogenomes) typically comprise a compact circular molecule that encodes the standard set of 37 genes, including 13 protein-coding genes (PCGs), 22 transfer RNA genes, and 2 ribosomal RNA genes.6 These genomes are widely valued for phylogenetics and species identification owing to their maternal inheritance, absence of introns, relatively high evolutionary rate, and highly conserved gene content across metazoans. In annelids, mitogenomes have proven particularly useful for resolving relationships at the family and genus levels, revealing gene-order variations in certain lineages, and facilitating comparative analyses of evolutionary patterns.7,8 However, gene-order conservation is generally high in Annelida, with most rearrangements occurring in specific clades, and mitogenomic data often provide stronger signal for shallower phylogenetic nodes than for deep splits.9 Recent systematic studies have substantially expanded annelid mitogenome datasets, yet taxon sampling remains highly uneven, with many families, including the ecologically important Onuphidae, still severely underrepresented.
Within Onuphidae, complete mitogenomes are particularly scarce. To date, only a few have been fully characterized, primarily from Diopatra species, which severely constrains robust phylogenetic assessments of intrafamilial relationships.5,10 Previous molecular phylogenies of Onuphidae, based on nuclear and mitochondrial markers, have revealed complex relationships, supported the monophyly of the subfamilies Onuphinae and Hyalinoeciinae, and suggested the presence of potential cryptic diversity within several genera.10,11 Mitogenome-level data could therefore provide additional resolution for closely related taxa, while also revealing evolutionary signals in gene content, nucleotide composition bias, and codon usage.12 In Onuphidae, several key questions remain unresolved, including the precise intergeneric relationships within subfamilies such as Hyalinoeciinae and the phylogenetic placement of Hyalinoecia relative to other onuphid lineages.
Hyalinoecia robusta is an ideal candidate for mitogenome characterization given its significant ecological role in deep-sea soft-sediment habitats, its distinctive morphology and epibenthic crawling behavior within the subfamily Hyalinoeciinae, and the taxonomic ambiguities surrounding onuphid diversity.2,10 The scarcity of genomic resources for this species and the subfamily Hyalinoeciinae contributes to broader challenges in understanding onuphid evolution, including potential cryptic speciation and physiological adaptations to deep-sea conditions. By generating and analyzing the complete mitogenome of H. robusta, this study addresses a clear and longstanding knowledge gap in annelid mitochondrial genomics. In this study, we assembled and annotated the complete mitochondrial genome of H. robusta and evaluated its phylogenetic position within Onuphidae using concatenated sequences of the 13 mitochondrial protein-coding genes. Our objectives were to characterize the genomic features of the H. robusta mitogenome and to provide a preliminary phylogenetic framework for this species based on currently available annelid mitochondrial genomes. The newly generated mitogenome also offers a valuable genetic resource for future molecular identification, comparative mitogenomic analyses, and biodiversity assessment of deep-sea benthic annelids.
Materials and Methods
Sample collection and DNA extraction
Specimens of Hyalinoecia robusta were collected from a single station in the South China Sea (17.29°N, 110.82°E) using a grab sampler during a research cruise on 6 April 2025, at a water depth of 1,627 m. The sampling site was characterized by soft, muddy sediment with a bottom water temperature of 2.73°C. A total of nine specimens was obtained from the sediment samples (Figure 1). All specimens were immediately preserved in 95% ethanol on board and transported to the laboratory under chilled conditions. One individual was morphologically identified as H. robusta based on key taxonomic characters. The voucher specimen (Holotype: TIO-BTS-ONU-2201) was deposited in the Third Institute of Oceanography, Ministry of Natural Resources, Xiamen, China. Total genomic DNA was extracted from muscle tissue using a standard cetyltrimethylammonium bromide (CTAB) protocol. The quantity and quality of the DNA were checked on a standard agarose gel electrophoresis and a Qubit Fluorometer (Invitrogen, USA), respectively.

Sequencing, assembly, and annotation
To obtain the complete mitochondrial genome sequence, approximately 1 μg of high-molecular-weight genomic DNA was used to construct a paired-end library. The DNA was purified using the TIANgel Midi Purification Kit (Tiangen Biotech Co., Ltd., Beijing, China) according to the manufacturer’s instructions. The purified DNA was then fragmented to an average insert size of 350 bp using a Covaris M220 sonicator. Sequencing libraries were prepared using the NEBNext Ultra™ DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s protocol. After adapter ligation, libraries were amplified with 8 PCR cycles. Library quality and insert size were validated using agarose gel electrophoresis and a Qubit Fluorometer before sequencing on the Illumina NovaSeq 6000 platform. Raw reads were trimmed to remove low-quality bases and adapter sequences using fastp with the following parameters: -q 10 -u 50 -y -g -Y 10 -e 20 -l 100 -b 150 -B 150.13 Approximately 6 Gb of Illumina short reads (clean data) were generated, yielding an average mitogenome coverage of >1,200×, with mitochondrial reads comprising about 0.8–1.2% of total reads.
All the clean reads were used for de novo assemblies using GetOrganelle v1.7.7.0 with the parameter: -R 10 -k 21,45,65,85,105 -F animal_mt.14 Protein-coding genes were initially annotated using MitoZ v3.6 and then manually checked by comparison with related annelid mitochondrial genomes available in GenBank.15 Transfer RNA genes and ribosomal RNA genes were identified using the MITOS web server with the invertebrate mitochondrial genetic code.16 Gene boundaries, start and stop codons were manually verified and refined by comparing the sequences with closely related onuphid and eunicid mitogenomes (e.g., Diopatra and Marphysa species) using MEGA X.17 Overlapping regions between adjacent genes were also checked visually to ensure annotation accuracy. Base composition and relative synonymous codon usage (RSCU) were calculated using the PhyloSuite v1.2.3.18 The AT and GC skews were calculated according to the following formulae: AT-skew = (A−T)/(A+T) and GC-skew = (G−C)/(G+C) (Perna and Kocher. 1995). The mitogenome of H. robusta was visualized using OGDRAW v1.3.1 with default parameters.19
Phylogenetic analysis
To elucidate the phylogenetic position of H. robusta within the order Eunicida, phylogenetic analyses were conducted using a concatenated mitochondrial dataset comprising 13 shared PCGs from 7 species within Eunicida, with Cryptonome barbada used as the outgroup. The nucleotide sequences of the 13 PCGs were extracted, aligned using MAFFT v7.310,20 and trimmed with TrimAl v1.4 prior to concatenation.21 The aligned genes were then concatenated into a single nucleotide dataset. In parallel, the corresponding amino-acid sequences translated from the same 13 PCGs were also concatenated into a protein dataset. The optimal substitution model was identified using ModelFinder v2.2.0 based on the Bayesian Information Criterion (BIC).22 Maximum-likelihood phylogenetic analyses were performed using IQ-TREE v2.2.0 with 1,000 ultrafast bootstrap replicates.23 We used iTOL v5 (https://itol.embl.de) to visualize the derived ML trees,24 with the nucleotide-based topology as the main tree and support values from both the nucleotide and amino-acid analyses were displayed together at corresponding nodes.
Results
Mitogenome organization of H. robusta
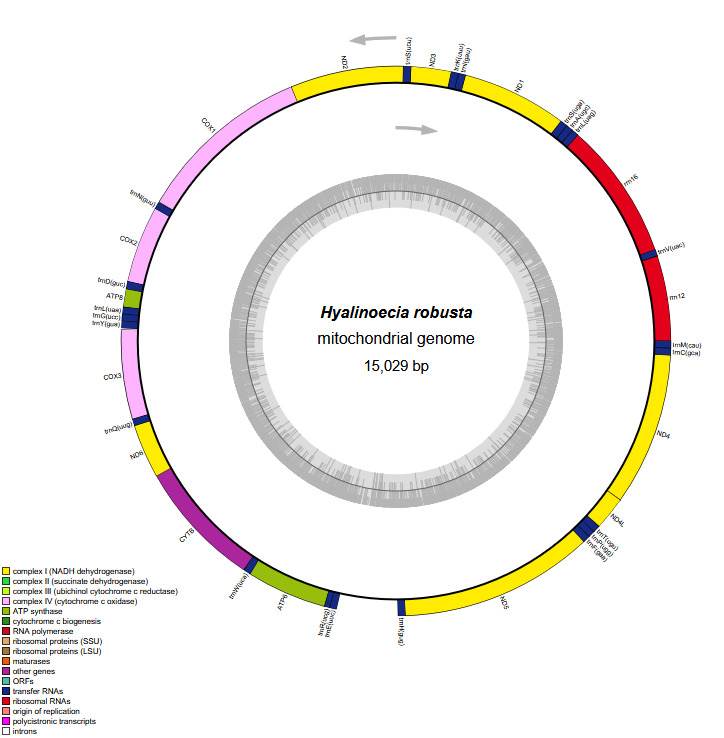
The complete mitogenome of H. robusta is a circular molecule of 15,029 bp in length (GenBank accession number PP790749) (Figure 2), which is longer than that of Diopatra sp. but shorter than those of other Eunicidae species (Table 1). The total length of intergenic regions is 526 bp, constituting 3.50% of the mitogenome, which is comparable to that observed in other Eunicida species, reflecting a compact mitochondrial genome organization in this order. The mitogenome of P. amurensis contains the standard set of 37 genes, including 13 PCGs, 22 transfer RNA genes, and 2 ribosomal RNA genes, together with a putative control region of 541 bp. All genes are encoded on the heavy strand.
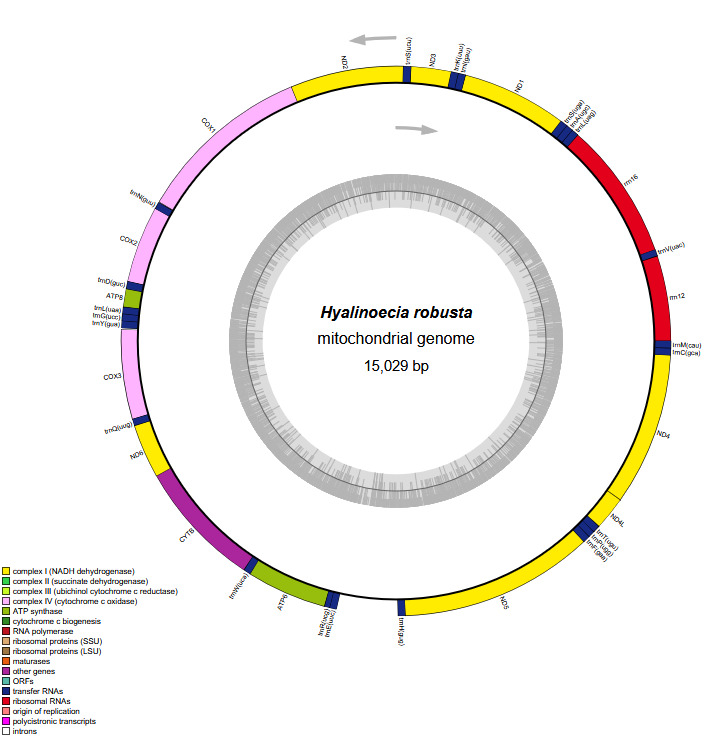
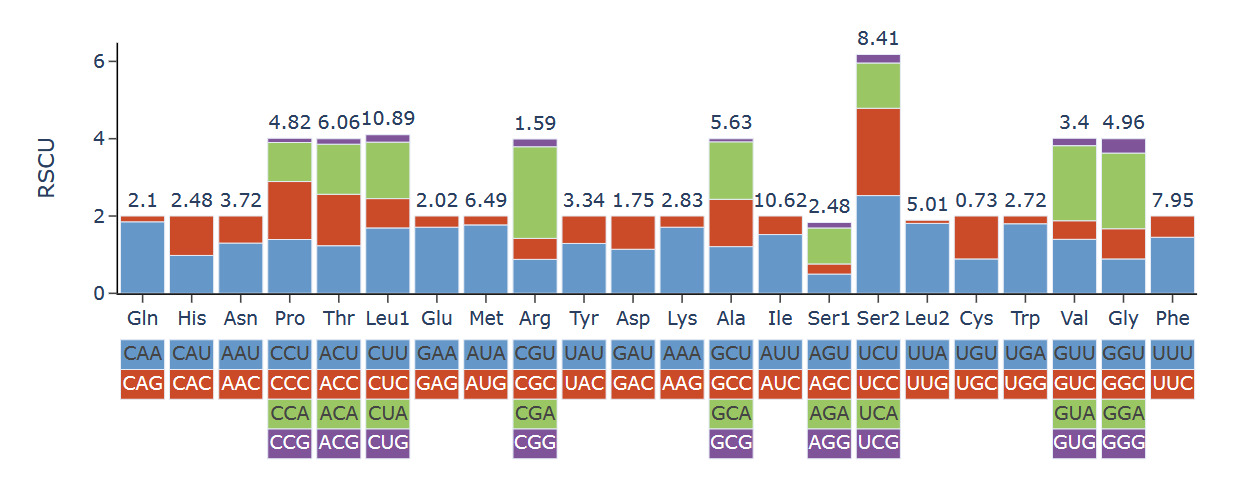
The mitogenome exhibited a nucleotide composition of 30.92% A, 35.53% T, 21.42% C, and 12.13% G, yielding an A+T content of 66.45%, which significantly exceeded the C+G content (33.55%; Table 1). Interestingly, the GC content of three Eunicidae species (H. robusta, Diopatra sp., Diopatra cuprea) was significantly lower than that of species from other families. The H. robusta mitogenome showed a clear bias in nucleotide composition with a negative AT skew (-0.069) and a negative GC skew (-0.277). The base composition and skewness are consistent with most studies in polychaete annelids. The RSCU analysis of the PCGs in the H. robusta mitogenome revealed that 31 codons have RSCU values greater than 1.00, among which 31end with either A or U, suggesting a strong AT bias in the third codon (Figure 3)
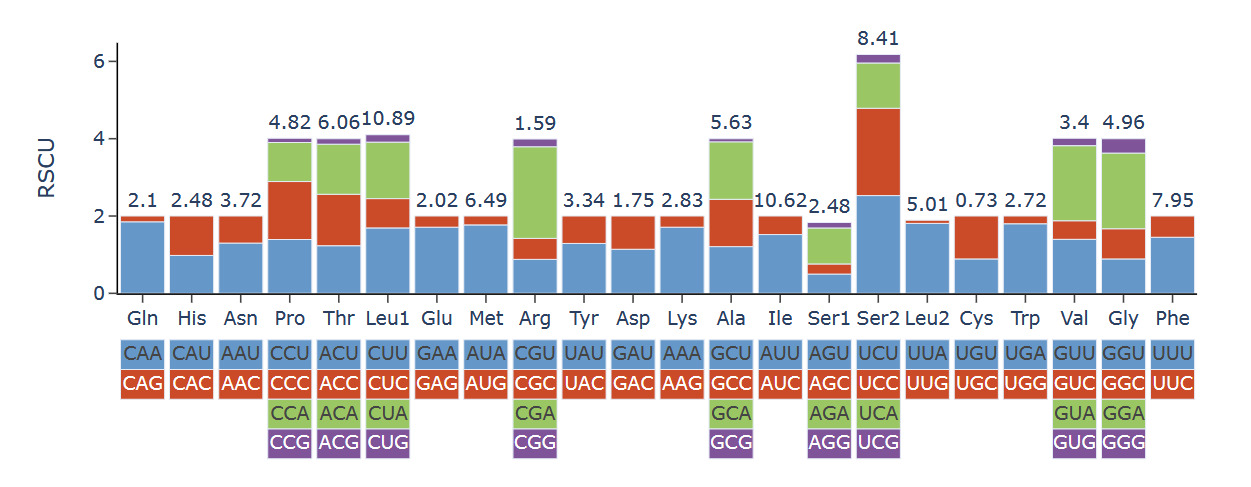
All PCGs encoded by the H. robusta mitogenome have a methionine (ATG) as the start codon. Four PCGs stop with codon “TAG” (namely, ND3, ND4, COX3, and CYTB), while the others stop with codon “TAA” (Table 2). Additionally, a 39 bp overlap is present between rrn16 and trnL(uag), an 8 bp overlap between ND6 and CYTB, and a 7 bp overlap between ND4L and ND4 in the H. robusta mitogenome.
Preliminary phylogenetic assessment
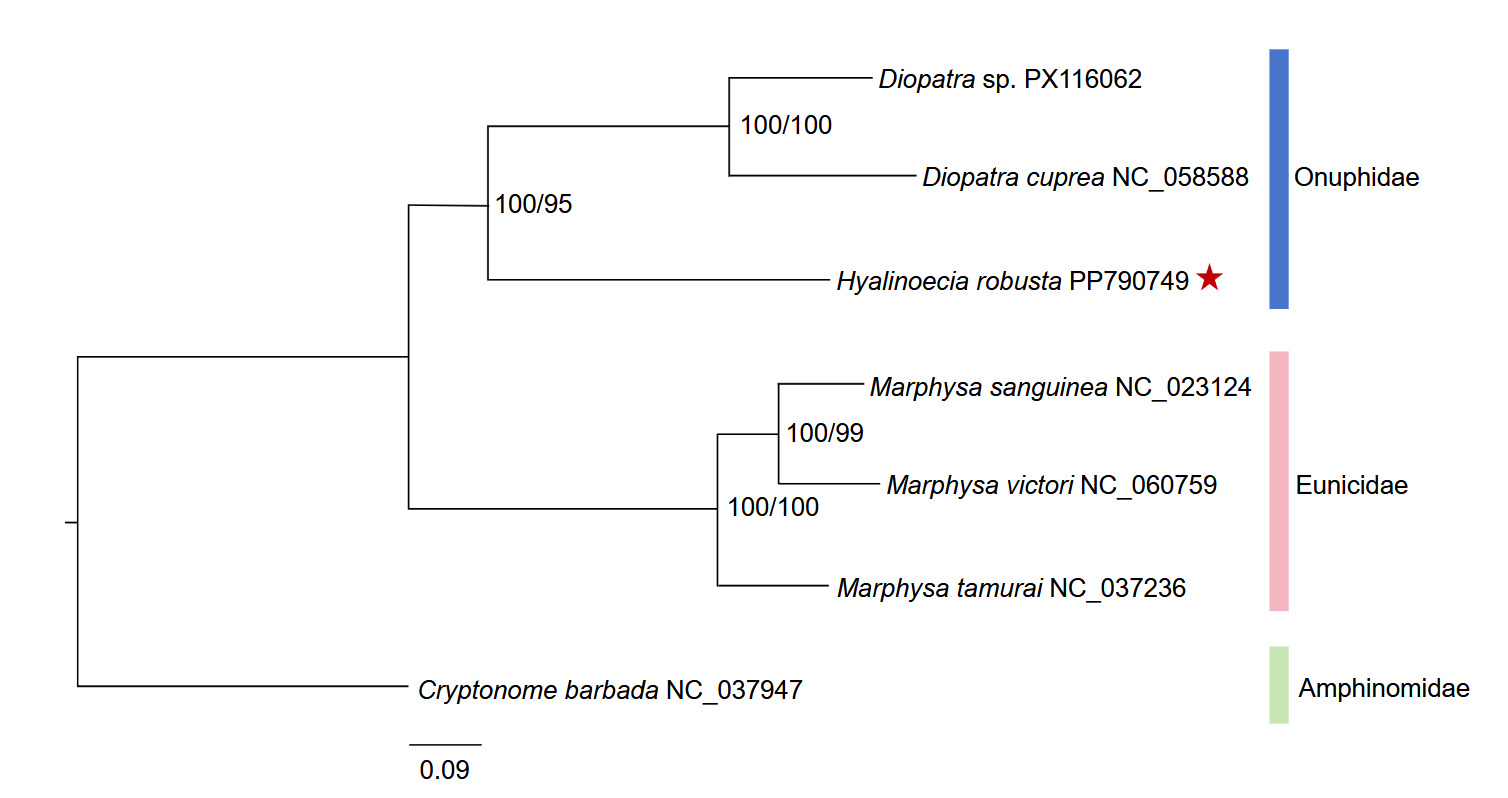
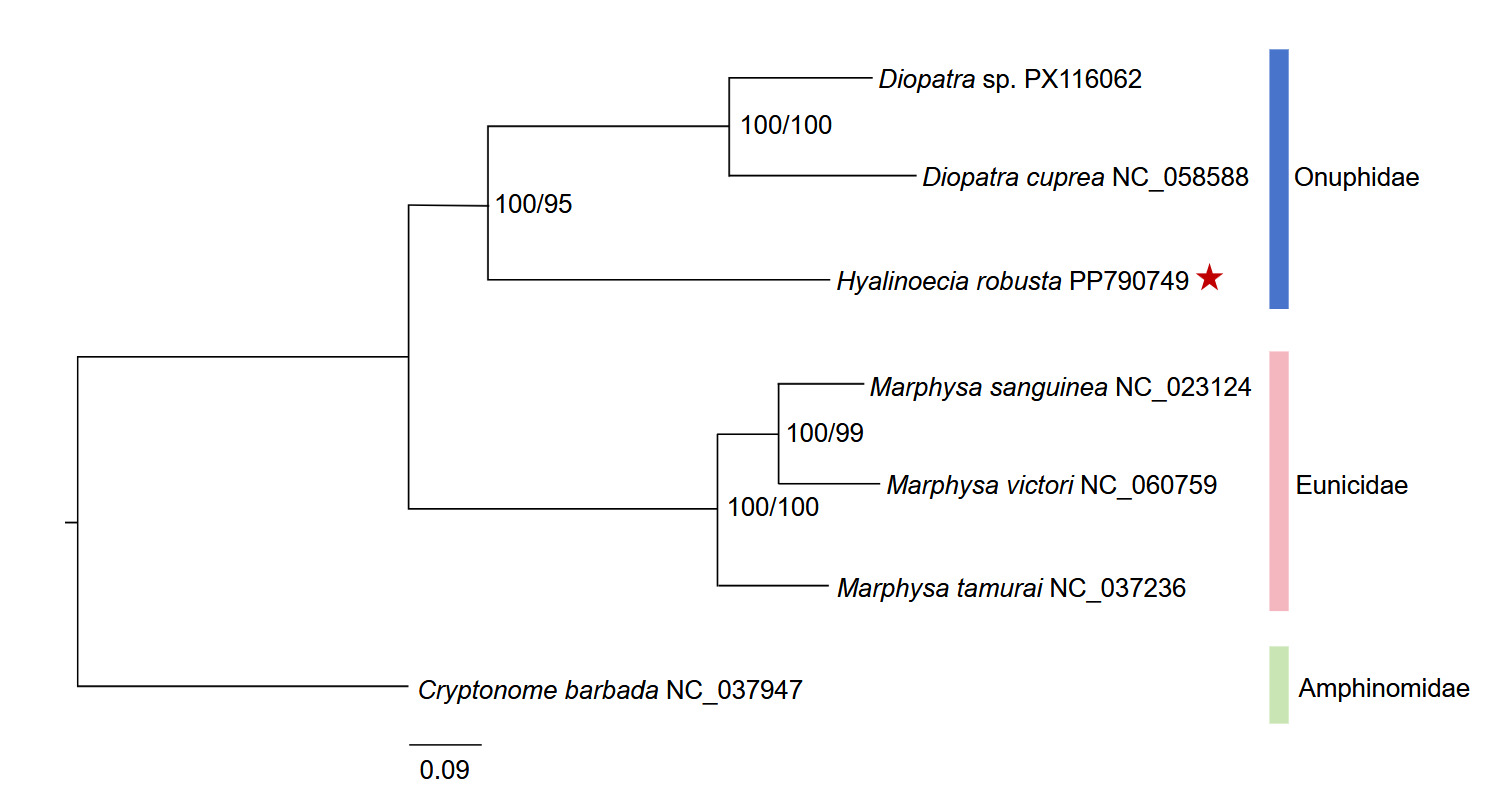
Phylogenetic reconstruction based on the concatenated 13 mitochondrial protein-coding genes yielded a consistent major topology across the nucleotide and amino-acid datasets (Figure 4). The tree was rooted with C. barbada. The ingroup was clearly divided into two highly supported clades corresponding to Eunicidae (three Marphysa species) and Onuphidae (H. robusta and two Diopatra species) (Figure 4). Within the onuphid clade, Diopatra sp. and D. cuprea formed a sister pair with maximal support (100/100), and H. robusta was recovered as their sister with strong support (100/95). Within the eunicid clade, M. sanguinea and M. victori grouped together (100/99), and this subclade was sister to M. tamurai (100/100). These results indicate that H. robusta is more closely related to Diopatra than to Marphysa within the present taxon sampling. The concordant clustering pattern obtained from the nucleotide and amino-acid datasets, together with the high node support values from the topology, supports the preliminary phylogenetic placement of H. robusta within the currently sampled onuphid taxa.
Discussion
In this study, we assembled and characterized the complete mitogenome of H. robusta, a deep-sea onuphid annelid, and performed a preliminary phylogenetic analysis based on concatenated mitochondrial PCGs. The H. robusta mitogenome is a circular molecule of 15,029 bp containing the standard set of 37 metazoan mitochondrial genes (13 PCGs, 22 tRNAs, and 2 rRNAs). Its overall organization, compact size, and gene content are highly consistent with those reported for other annelids, particularly members of Eunicida.5,12,25 The total length falls within the range observed in Onuphidae (14,985-15,050 bp; Table 1) and is slightly shorter than that of most Eunicidae species, which typically exceed 15,150 bp. The intergenic region length (526 bp, 3.50% of the genome) is comparable to that of Diopatra sp. and D. cuprea,5,12 reflecting a generally compact mitogenome architecture in this family.
The nucleotide composition of the H. robusta mitogenome exhibits a marked A+T bias (66.45%), with negative AT skew (-0.069) and GC skew (-0.277). Such strong AT enrichment is a common feature in annelid mitochondrial genomes, especially in polychaetes.7 More interestingly, when comparing across Eunicida, the three Onuphidae species (H. robusta, Diopatra sp., D. cuprea) show significantly lower GC contents (30.25-33.55%) than the representatives of Eunicidae (39.04-40.94%) and the outgroup Cryptonome barbada (43.23%). This consistent difference suggests that Onuphidae may have experienced distinct evolutionary pressures that have shaped their mitochondrial base composition. Lower GC content is often associated with reduced energetic costs for replication and transcription. It could reflect adaptation to specific environmental conditions, such as hypoxic or deep-sea habitats where energetic efficiency is at a premium.26 However, the precise ecological or molecular mechanisms driving this GC disparity among eunicidan families remain to be explored. The RSCU analysis further supports the strong A+T bias, as the majority of codons with RSCU > 1 end with A or U, a pattern typical of AT-rich annelid mitogenomes.
Phylogenetic analyses based on concatenated nucleotide and amino acid sequences of the 13 mitochondrial PCGs consistently recovered H. robusta as the sister group to the two Diopatra species, with strong bootstrap support (100/95 for the nucleotide and amino acid datasets, respectively). The three Marphysa species (family Eunicidae) formed a separate, well-supported clade. This topology aligns with the current taxonomic recognition of Onuphidae and Eunicidae as distinct families within Eunicida,10 and provides preliminary mitogenomic evidence for a close affinity between Hyalinoecia and Diopatra. Previous phylogenetic studies employing nuclear and mitochondrial markers (16S and 18S rDNA) also supported the monophyly of Onuphidae and placed Hyalinoecia within a clade containing Diopatra and other onuphid genera.10 Thus, our results offer independent mitogenomic support for these findings and extend the currently limited mitogenomic framework for Onuphidae, which previously included only Diopatra species.5,12
The phylogenetic interpretation of the present dataset should remain cautious. Complete mitogenomes are still scarce for Onuphidae, and taxon sampling within the family remains sparse. Previous phylogenetic studies based on nuclear and mitochondrial markers have shown that onuphid relationships are more complex than can be fully resolved with a few representative taxa alone.27 In the present study, both nucleotide- and amino-acid-based analyses recovered the same major clustering pattern, which supports the robustness of the principal relationships observed here. However, slight differences in node support between the two datasets suggest that deeper relationships among the sampled lineages are not yet fully stabilized. Accordingly, the phylogeny presented here is best regarded as a preliminary assessment of the placement of H. robusta, rather than a comprehensive reconstruction of intrafamilial relationships within Onuphidae. Beyond its phylogenetic implications, the mitogenome reported here also represents a useful genetic resource for marine annelid research. Onuphid worms are ecologically important members of benthic communities and, in some cases, are relevant to aquaculture and bait-worm use. Additional mitochondrial genome data from this group will be valuable for molecular identification, comparative mitogenomics, and biodiversity assessment of marine benthic annelids. Future studies incorporating broader taxon sampling, additional onuphid mitogenomes, and nuclear genomic markers will be necessary to test the stability of the relationships recovered here and to establish a more complete phylogenetic framework for Onuphidae and related annelid lineages.
Conclusion
This study reports the complete mitochondrial genome of H. robusta, a marine annelid of the family Onuphidae. The mitogenome is 15,029 bp in length and contains the typical set of 37 mitochondrial genes, with a pronounced A+T bias of 66.45%. Phylogenetic analyses based on concatenated nucleotide and amino-acid sequences of the 13 mitochondrial PCGs consistently support a close relationship between H. robusta and Diopatra species. The newly characterized mitogenome provides a useful genetic resource for molecular identification and future comparative studies of marine annelids.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (42006127), the Science & Technology Fundamental Resources Investigation Program (Grant No.2024FY101000), the Basic Science Research Fund of the Third Institute of Oceanography, MNR (No. TIO2016043), the National Special Project on GasHydrate of China (Nos. DD20221706 and No. DD20230065).
Authors’ Contribution
Formal Analysis: Yaqin Huang (Equal), Site Luo (Equal), Heshan Lin (Equal). Funding acquisition: Yaqin Huang (Equal), Xuebao He (Equal). Methodology: Yaqin Huang (Lead). Project administration: Yaqin Huang (Equal), Xuebao He (Equal). Writing – original draft: Yaqin Huang (Lead). Investigation: Xuebao He (Equal), Junhui Lin (Equal), Kun Liu (Equal), Jianfeng Mou (Equal). Resources: Qianyong Liang (Lead). Supervision: Zhongbao Li (Lead). Writing – review & editing: Zhongbao Li (Equal).
Competing of Interest – COPE
No competing interests were disclosed.
Ethical Conduct Approval – IACUC
The sample collection and animal experiments adhered to regulations and guidelines for the care and use of laboratory animals, and received approval from the Animal Care and Use Committee of Third Institute of Oceanography, Ministry of Natural Resources (protocol code TIO-IACUC-01-2023-03-20).
Informed Consent Statement
All authors and institutions have confirmed this manuscript for publication.
Data Availability Statement
All are available upon reasonable request.