Introduction
Cyprinidae is divided into 12 subfamilies and includes 379 genera. The cyprinid genus Acrossocheilus Oshima, 1919 belongs to the Barbinae, which includes at least 27 genera. The distribution of Acrossocheilus is restricted mainly to South China and the northern part of Vietnam and Laos, and the genus is especially diverse in China (with 21 species).1 Acrossocheilus species occur in tropical, subtropical, and temperate regions,2 and they are commonly found in mountain streams and rivers, including freshwater habitats in the middle and lower reaches of drainage systems in Fujian, Anhui, Zhejiang, and other central and southern Chinese provinces.3
Species of Acrossocheilus are generally identified by morphological characters. To date, 26 species have been recognized in Acrossocheilus1,4: A. iridescens, A. microstoma, A. longipinnis, A. jishouensis, A. yalyensis, A. aluoiensis, A. baolacensis, A. lamus, A. macrophthalmus, A. malacopterus, A. monticola, A. multistriatus, A. parallens, A. rendahli, A. spinifer, A. wenchowensis, A. wuyiensis, A. xamensis, A. yunnanensis, A. paradoxus, A. hemispinus, A. ikedai, A. fasciatus, A. clivosius, A. beijiangensis, and A. kreyenbergii. The genus can be tentatively divided into two species groups: a striped group, characterized by a plain or inconspicuous dark stripe along the lateral line of the flank, and a barred group, characterized by five to eight vertical black bars along the flank.4 However, ontogenetic change and sexual dimorphism can affect body coloration, lateral stripes or bars, dorsal-fin serrations, mouth and lip morphology, and meristic characters; therefore, the systematics of the two groups remains unresolved. Originally, the barred group was referred to as Acrossocheilus sensu stricto, whereas the striped group was affiliated with Poropuntius.5
In China, the striped group includes A. ikedai, A. xamensis, A. cf. xamensis, A. yunnanensis, and A. malacopterus (a species formerly misidentified as A. elongatus).2 In contrast, the barred group includes A. beijiangensis, A. clivosius, A. fasciatus; A. hemispinus, A. iridescens, A. jishouensis; A. kreyenbergii, A. lamus, A. longipinnis, A. microstoma, A. monticola, A. paradoxus, A. parallens, A. spinifer, A. wenchowensis, and A. wuyiensis and it is mostly distributed in South China. The exception is A. lamus, which is endemic to Vietnam.6 Identification based only on morphology can be difficult, especially when key diagnostic traits such as coloration, bars, stripes, and serrations on the unbranched dorsal-fin ray change during the transition from juvenile to adult stages. Consequently, the distribution of narrowly endemic species may be underestimated, and some GenBank accessions may reflect historical identifications that require voucher-based confirmation.
The taxonomic history of Acrossocheilus is marked by many contradictions, disagreements, and revisions. For example, A. iridescens was formerly classified as Cyclocheilichthys iridescens, Barbus barbodon, and B. paradoxus quinquefasciatus. Acrossocheilus iridescens subsp. yuanjiangensis was later described as A. microstoma, and A. iridescens subsp. zhujiangensis was later identified as A. longipinnis. Acrossocheilus stenotaeniatus was once considered synonymous with A. iridescens but has been treated as a junior synonym of A. longipinnis.7 In addition, A. kreyenbergii was formerly described as Gymnostomus kreyenbergii and as B. hemispinus subsp. cincta; in China, A. hemispinus has been recognized as distinct from A. cinctus, with the latter treated as a junior synonym of A. kreyenbergii.8
Morphological studies have also attempted to clarify relationships among Acrossocheilus species and closely related genera. Based on morphological diagnoses, A. elongatus was assigned to Onychostoma.9 Zacco platypus was treated as a synonym of A. parallens based on ontogenetic variation in body characteristics.10 Overall, uncertainty in Acrossocheilus taxonomy can be attributed partly to variation in morphological characters, limited voucher comparison, and the use of historically unstable names. Without definitive integrative diagnoses, synonyms, homonyms, and misidentifications may persist.
Molecular analyses may provide new insights into the systematics of Acrossocheilus because the taxonomic status of several nominal species is uncertain based on morphology alone. To resolve the taxonomic status of A. formosanus, A. labiatus, and A. fasciatus in Taiwan, horizontal starch-gel electrophoresis was used, confirming that the three nominal taxa were identical and should be reclassified as A. paradoxus.11 A prior study sequenced the mitochondrial ND4 gene from seven Chinese barred Acrossocheilus species to test their validity.12 That study supported a lineage comprising A. fasciatus, A. hemispinus, A. parallens, A. beijiangensis, A. spinifer, and A. hemispinus subsp. cinctus and proposed that A. hemispinus and A. hemispinus subsp. cinctus was a separate species.12 The mitochondrial control region was also used to examine relationships within the barred group; specimens previously identified as A. fasciatus and A. wuyiensis from Fujian Province were inferred to represent A. wuyiensis.6
Despite these efforts, most molecular studies have focused primarily on phylogenetic relationships, and explicit species-delimitation analyses within Acrossocheilus remain limited.13 In this study, we used published mitochondrial genomic data to address three specific objectives: (1) reconstruct the mitochondrial phylogeny of sampled Acrossocheilus and related Onychostoma taxa using the 13 mitochondrial protein-coding genes; (2) test whether the species complexes involving A. longipinnis-A. iridescens-A. barbodon, A. hemispinus-A. parallens, A. paradoxus, and A. beijiangensis-A. stenotaeniatus-A. spinifer are congruent with current morphology-based taxonomy; and (3) evaluate whether mitochondrial evidence supports reciprocal monophyly of Acrossocheilus and Onychostoma. Because only mitochondrial data are available for all included samples, we treat the resulting MOTUs as testable hypotheses for future integrative taxonomy rather than as final species-level decisions.
Materials and Methods
Data Collection
We downloaded 31 complete mitochondrial genomes representing 22 cyprinid species from the National Center for Biotechnology Information (NCBI) (Table 1). The dataset comprised 25 Acrossocheilus mitogenomes assigned to 16 sampled Acrossocheilus names in GenBank, three Onychostoma taxa, and three Spinibarbus outgroup taxa. The Onychostoma taxa were O. barbatulum (Pellegrin, 1908), O. meridionale (Kottelat, 1998), and O. gerlachi (Peters, 1881). Spinibarbus denticulatus (Oshima, 1926), S. hollandi (Oshima, 1919), and S. sinensis (Bleeker, 1871) were used as outgroups.
Because 26 species are recognized in Acrossocheilus in the Introduction, the sampling gap was made explicit. The recognized species not represented by complete mitogenomes in the present dataset were A. microstoma; A. yalyensis; A. aluoiensis; A. baolacensis; A. lamus; A. macrophthalmus; A. malacopterus; A. multistriatus; A. rendahli; A. xamensis; A. ikedai; and A. clivosius. Their absence may reduce the present dataset’s ability to detect additional lineages, test the full monophyly of species groups, and evaluate whether unsampled taxa would alter MOTU membership. Accordingly, all delimitation results are interpreted as hypotheses for the sampled taxa only.
For each complete mitochondrial genome, the 13 protein-coding genes (ATP6, ATP8, COX1, COX2, COX3, CYTB, ND1, ND2, ND3, ND4, ND4L, ND5, and ND6) were extracted according to GenBank annotations. Each gene was checked under the vertebrate mitochondrial genetic code to confirm reading-frame consistency and to screen for obvious annotation errors. Genes were aligned separately in MEGA v6.014 using the vertebrate mitochondrial genetic code and default settings unless otherwise specified. PhyloSuite v1.2.215 was used for sequence management, concatenation of the 13 aligned protein-coding genes, format conversion, and preparation of input files for partitioning and phylogenetic analyses.
Molecular Phylogenetics
We used Bayesian inference (BI) and maximum likelihood (ML) methods to estimate phylogenetic relationships using the concatenated dataset. The optimal partitioning strategy for the dataset was inferred using PartitionFinder v216 in PhyloSuite, under a greedy search algorithm with linked branch lengths based on the corrected Akaike Information Criterion (AICc). The general time-reversible model with gamma-distributed rate variation and a proportion of invariable sites (GTR+G+I) was selected as the most appropriate model for subsequent analyses. BI analyses were performed using MrBayes v3.2.6.17 20 million Markov Chain Monte Carlo (MCMC) generations were used, with tree sampling every 1,000 generations to calculate posterior probabilities (PPs). We used four chains and two independent runs, and the first 25% of generations were discarded as burn-in. Convergence was assessed by examining the average standard deviation of split frequencies and the effective sample size (ESS) for all parameters in Tracer v1.7.18 An ESS > 200 was used as a good indicator of convergence. ML analyses were conducted with the best partitioning scheme selected in PartitionFinder and run in IQ-TREE v1.6.819 independently ten times with 1,000 ultrafast bootstrap (BS) replicates to obtain branch support values. A BS value > 70 was considered to indicate strong support for a given clade in the ML analyses.
Molecular Species Delimitation
First, we used the Generalized Mixed Yule Coalescent (GMYC) model20 through the online server (https://species.h-its.org/gmyc/) to infer molecular clades based on the phylogeny. The GMYC analysis was conducted with the multiple-threshold version. This is a likelihood-based method for delimiting species by fitting within- and between-species branching models to reconstructed gene trees using an ultrametric tree. In this analysis, an ultrametric tree was obtained in BEAST v1.8.2.21 For the BEAST analysis, the Yule process of speciation and the uncorrelated log-normal relaxed clock model were used as tree priors. MCMC chains were run for 50 million generations with tree sampling every 5,000 generations. The first 10% of generations were discarded as burn-in.
Second, we used the Bayesian Poisson Tree Processes (bPTP) model22 through the online server (https://species.h-its.org/ptp/). This is an updated version of the original ML Poisson Tree Processes (PTP) model with Bayesian PPs, providing more accurate results. Compared with the GMYC model, the bPTP model is a more robust and simpler method. We used the ML tree as input and ran bPTP analyses for 500,000 MCMC generations, setting the thinning value at 100 and burn-in at 0.1.
Third, we used the Automatic Barcode Gap Discovery (ABGD) method23 through its online server (https://bioinfo.mnhn.fr/abi/public/abgd/). This is a distance-based method that relies on the gap in the distribution of interspecific and intraspecific genetic distances. The analyses were performed with the following settings: a prior limit to intraspecific diversity (P) from 0.001 to 0.1, a relative gap width (X) of 0.9, 20 steps, and the Jukes-Cantor (JC69) model.
Fourth, we used Bayesian Phylogenetics and Phylogeography (BPP) v4.3.824,25 to confirm the delimitation results from the above methods. This is a validation method that applies reversible-jump Markov Chain Monte Carlo (rjMCMC) to estimate the PPs for different hypotheses of species delimitation. The species recovered from the GMYC, bPTP, and ABGD analyses were used as putative species, yielding a total of 15 taxa to test. We used default settings with speciesdelimitation = 1, speciestree = 1, algorithm = 0, finetune = 2, usedata = 1, and cleandata = 0. The reversible-jump MCMC analyses were run for 100,000 generations and sampled every two generations, with 8,000 samples discarded as burn-in.
Results
Alignment Dataset
The final dataset consisted of 31 complete mitochondrial genomes listed in Table 1. The sequence lengths reported in Table 1 refer to complete mitochondrial genome lengths, not to the concatenated 13-gene alignment. The final alignment length, number of variable sites, number of parsimony-informative sites, and mean pairwise sequence divergence should be calculated directly from the final concatenated 13-PCG alignment before submission. These statistics cannot be derived reliably from the accession table or figure images alone; therefore, no numerical values are fabricated here.
Phylogenetic Relationships
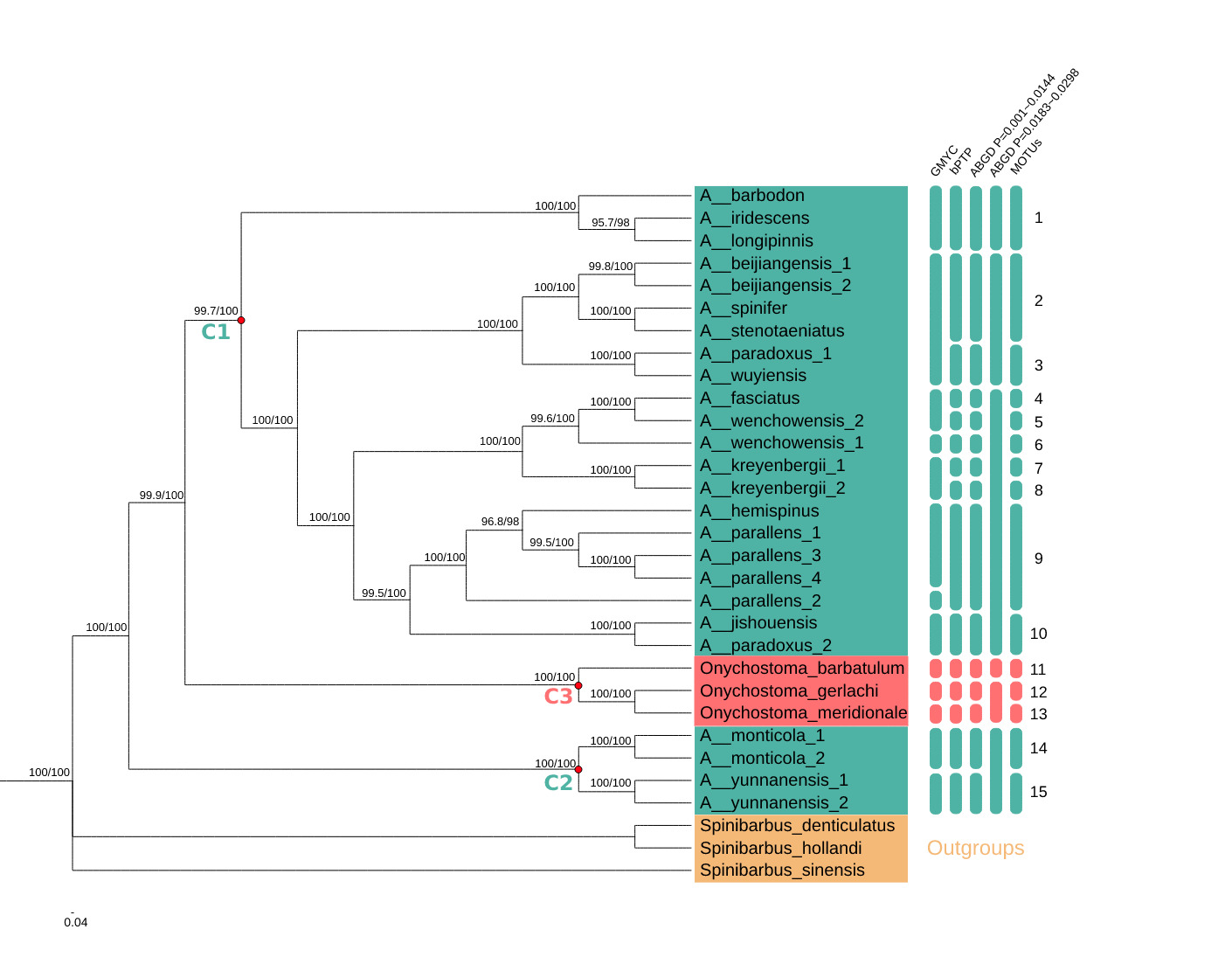
Phylogenetic analyses of the mitochondrial dataset using ML and BI approaches produced identical topologies with high BS support and PPs. Acrossocheilus was divided into two major clades, defined here as C1 and C2, that were supported with strong PPs and BS scores (100%) (Fig. 1).
The C1 clade was split into several well-supported subclades. A. longipinnis and A. iridescens grouped together and then clustered with A. barbodon into one clade with 1.00 PP and 100% BS support. A. hemispinus nested within a clade of A. parallens, and together these taxa were sister to a clade including one individual of A. paradoxus and A. jishouensis with 100% BS support and 0.995 PP. A. wenchowensis formed a paraphyletic group with the inclusion of A. fasciatus, and together these taxa were sister to a monophyletic group of A. kreyenbergii with 1.00 PP and 100% BS support. A monophyletic group of A. beijiangensis formed a clade with A. stenotaeniatus and A. spinifer, which was sister to a clade comprising A. wuyiensis and another individual of A. paradoxus with 100% BS support and 1.00 PP.
The C2 clade included only two species, each comprising two individuals: A. monticola and A. yunnanensis. Both species were monophyletic with 100% BS support and 1.00 PP. Notably, the genus Onychostoma formed a well-supported clade (100% BS and 1.00 PP), defined here as C3, which was nested between the two Acrossocheilus clades (C1 and C2) (Fig. 1). The clades C1 and C3 were more closely related and together were sister to C2.
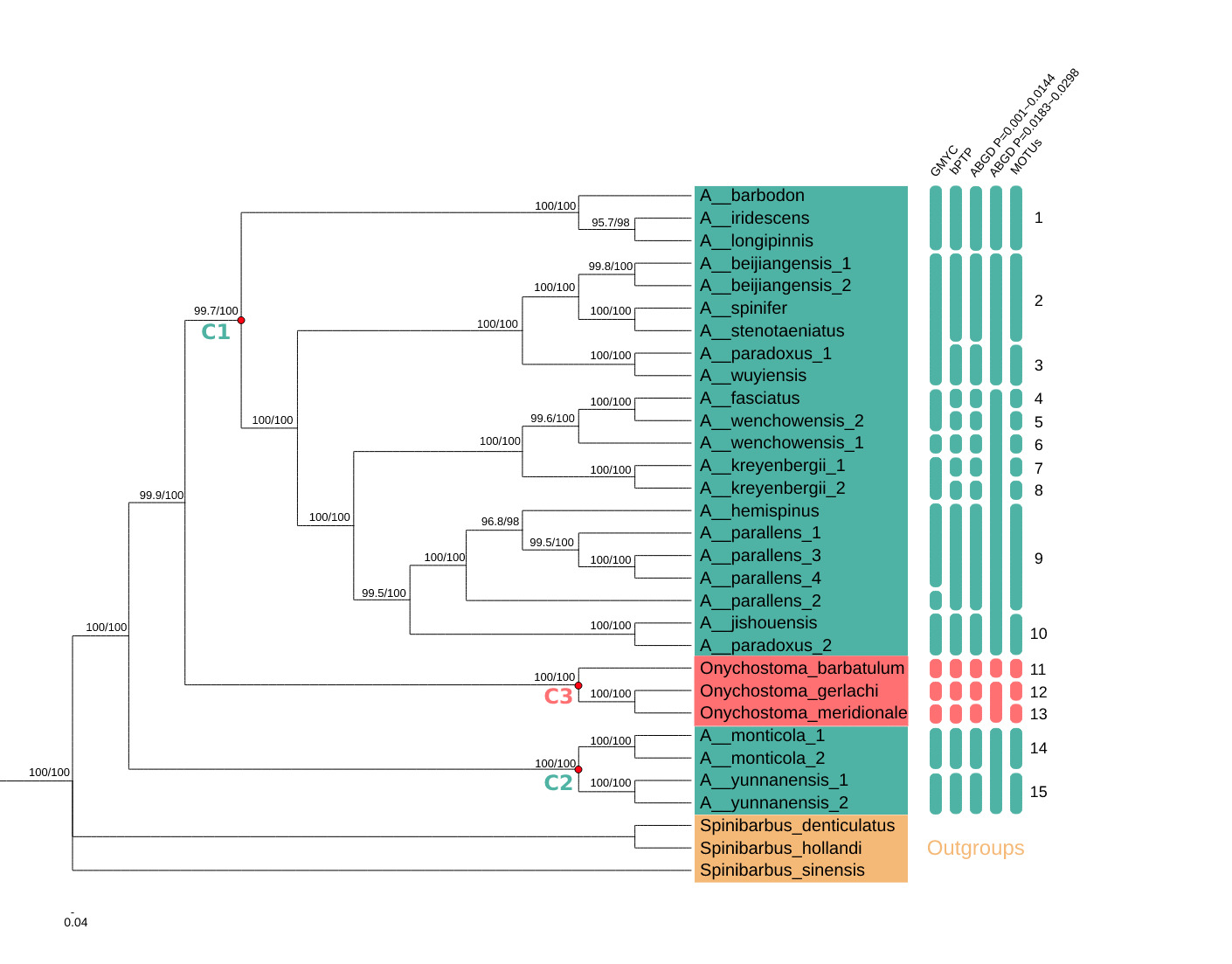
Species Delimitation
Across sampled Acrossocheilus and Onychostoma lineages, the number of inferred molecular operational taxonomic units (MOTUs) varied from six to 15 depending on the method and parameter settings (Fig. 1). The multiple-threshold GMYC analysis estimated 13 putative species, with a confidence interval of four to 19 and a branching-pattern transition at -0.8592776. The bPTP analysis estimated 15 putative species; the highest Bayesian-supported solution was congruent with the ML solution, with an acceptance rate of 0.29049, 50,103 merge events, and 49,897 split events.
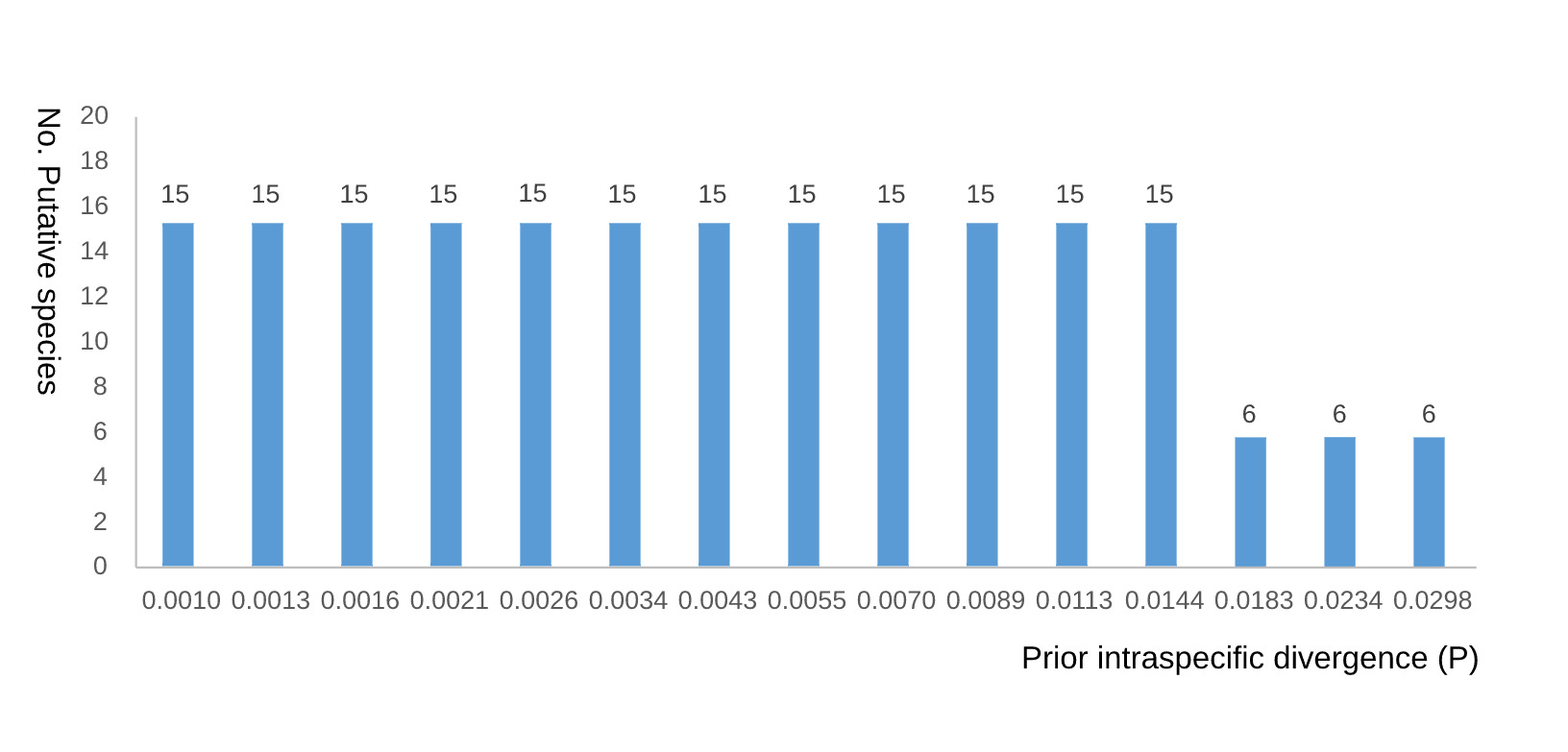
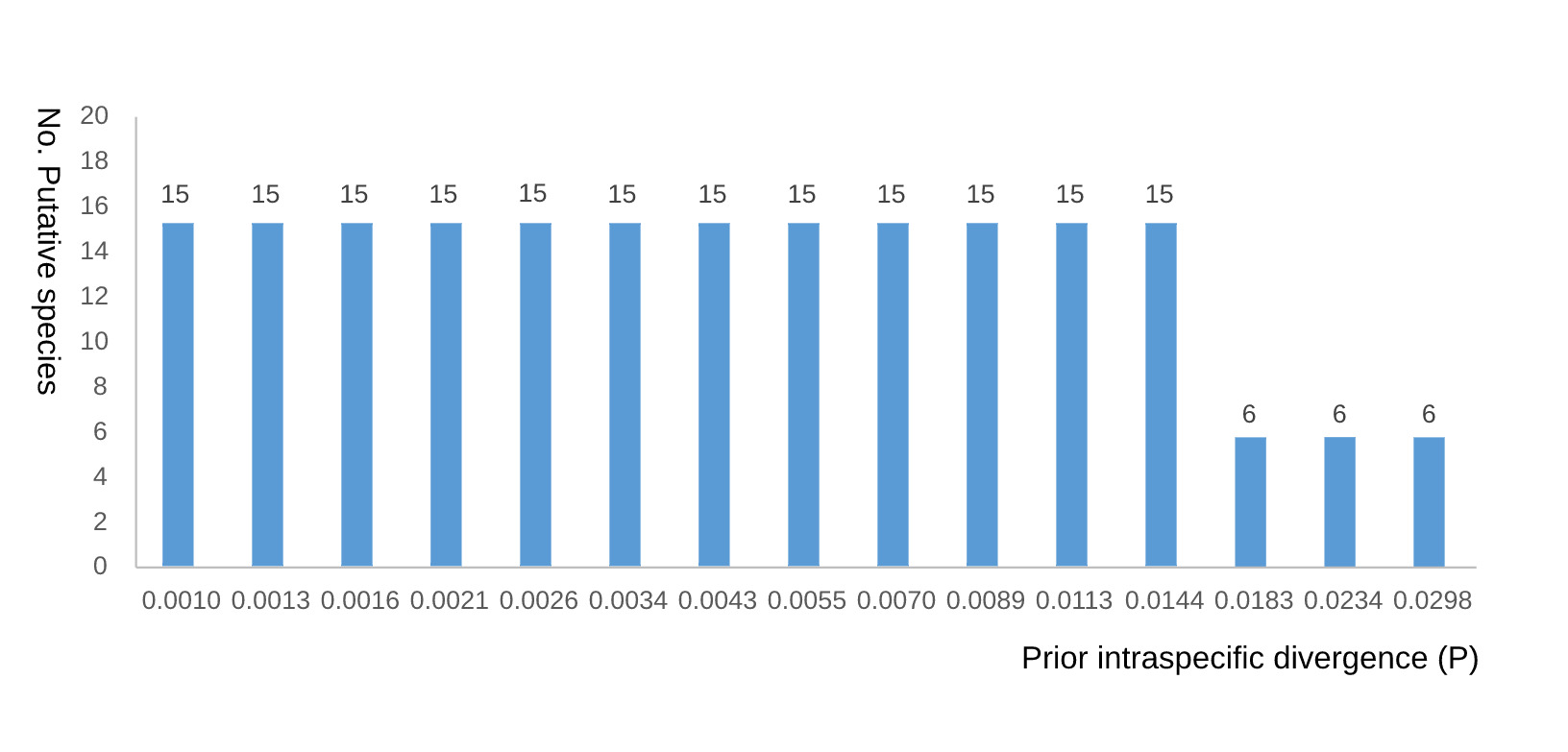
The ABGD results were sensitive to the prior limit of intraspecific divergence (P). Fifteen putative species were recovered at P = 0.0010, 0.0013, 0.0016, 0.0021, 0.0026, 0.0034, 0.0043, 0.0055, 0.0070, 0.0089, 0.0113, and 0.0144, matching the bPTP result. Six putative species were recovered at P = 0.0183, 0.0234, and 0.0298 (Fig. 2). Thus, the six-species ABGD result is reported together with its P-value interval rather than as an isolated conclusion.
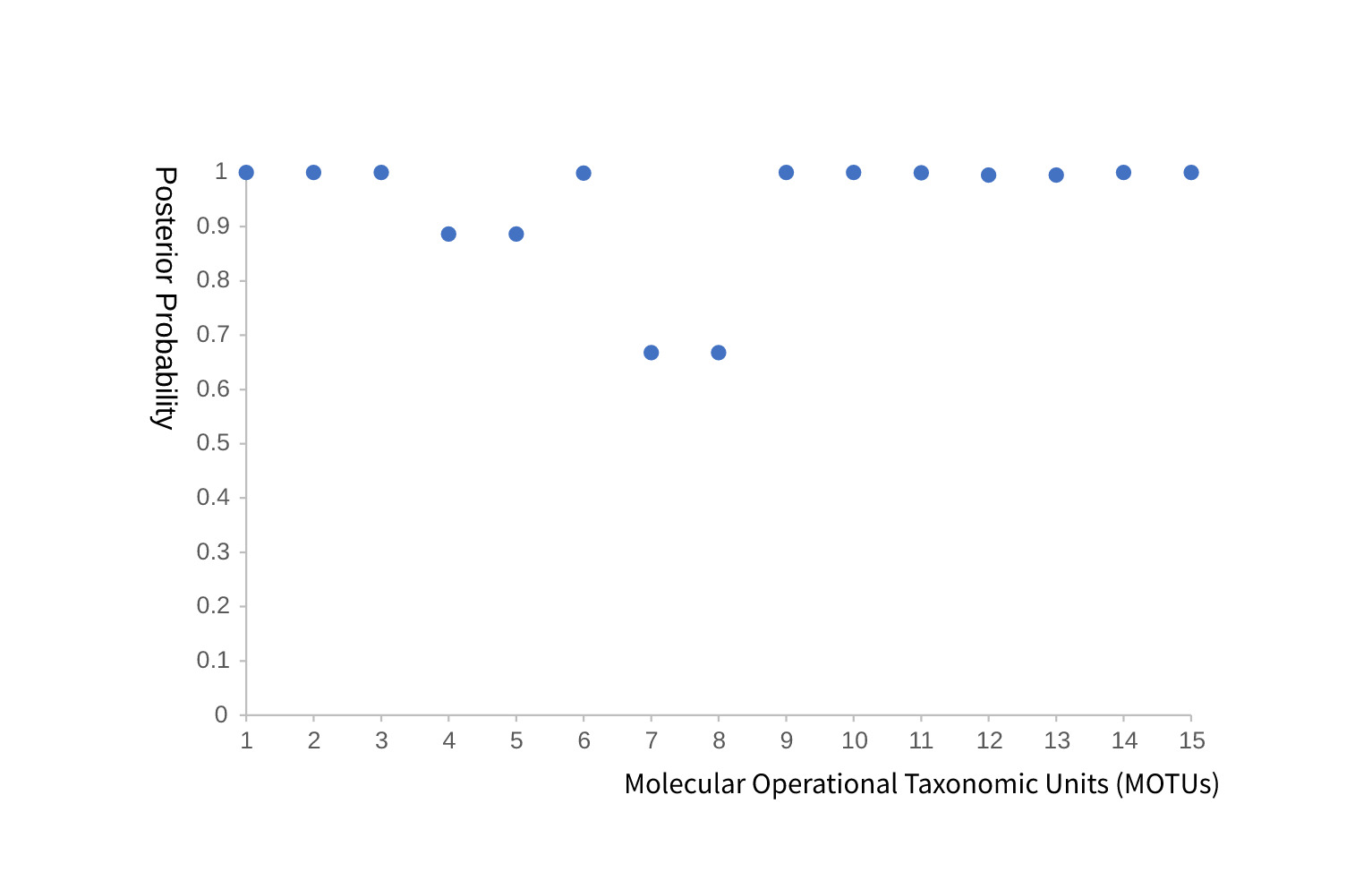
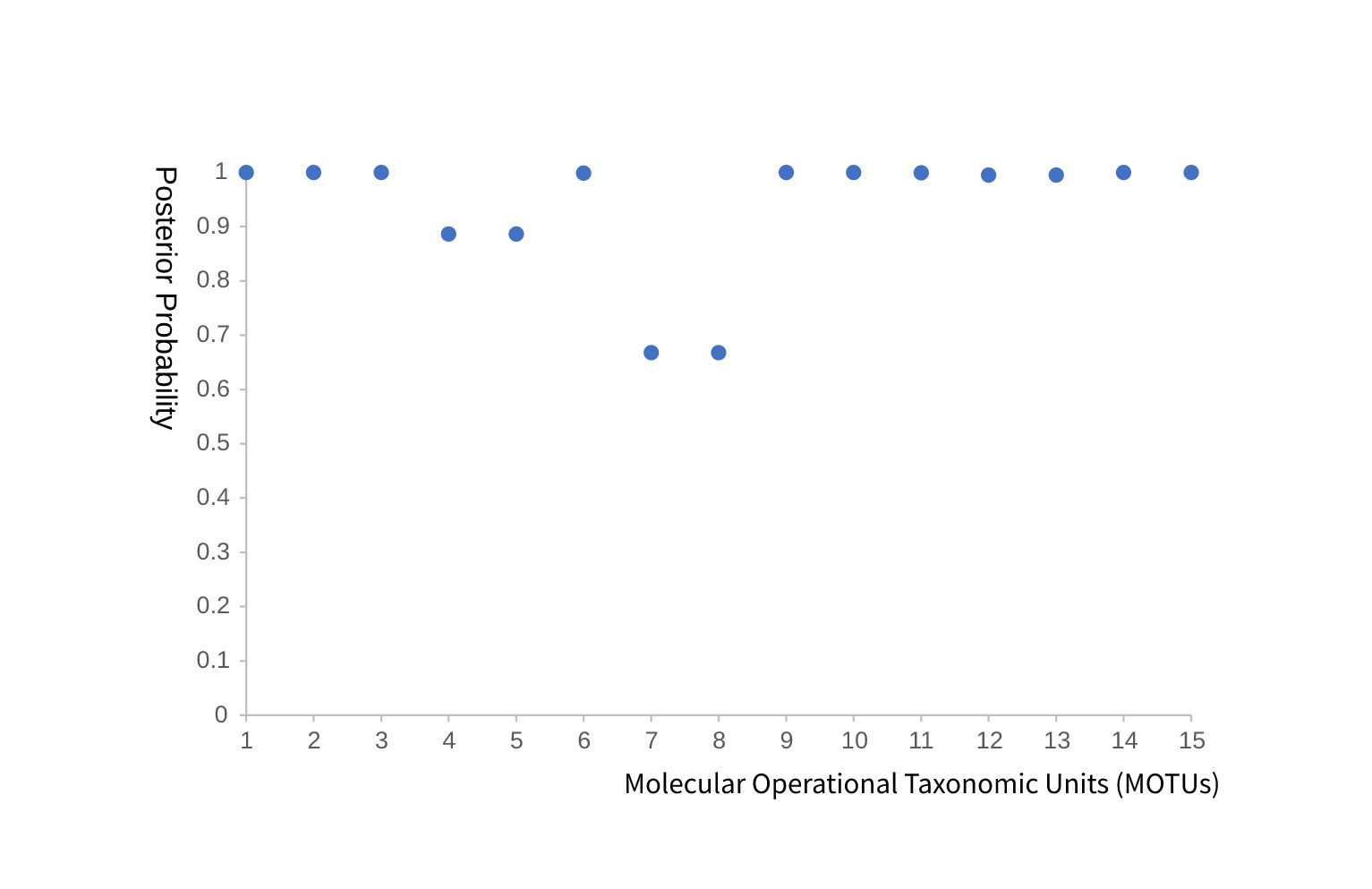
The BPP analysis of the 15 candidate MOTUs showed that 11 MOTUs had posterior probabilities greater than 0.95. The four remaining MOTUs, corresponding to MOTUs 4, 5, 7, and 8 in Fig. 3, received lower support, ranging from approximately 0.66 to 0.88. These moderately supported units should therefore be regarded as provisional mitochondrial hypotheses that require validation with nuclear markers and morphology.
Discussion
Overall, the four molecular species-delimitation methods yielded broadly congruent results but were not identical. bPTP and ABGD were highly consistent when ABGD P values ranged from 0.0010 to 0.0144, both of which supported 15 candidate MOTUs. GMYC recovered 13 putative species and therefore provided partial support for the same general pattern, although GMYC can overestimate species numbers when single-locus branching patterns are uneven.26 ABGD yielded only six putative species under higher P values (0.0183-0.0298), emphasizing that distance-based delimitation is sensitive to prior assumptions about intraspecific divergence. We therefore interpret the 15-MOTU scheme not as a final taxonomy, but as the most informative working hypothesis for the present mitochondrial dataset.
The proposed MOTUs highlight several groups requiring taxonomic reassessment. A. longipinnis, A. iridescens, and A. barbodon formed a strongly supported mitochondrial clade and were assigned to the same MOTU under the most congruent delimitation settings. Previous morphology-based work treated A. longipinnis as a senior synonym of A. stenotaeniatus.7 Rather than concluding that A. longipinnis is invalid, the present results indicate that A. longipinnis may be closely related to, recently diverged from, introgressed with, or misidentified relative to A. iridescens and A. barbodon. Additional nuclear loci, topotypic material, and direct examination of diagnostic characters are required before any synonymy is formally proposed. Earlier catalogue-level synonymy treatments should therefore be regarded as taxonomic hypotheses for integrative testing rather than definitive evidence.27
The A. beijiangensis-A. stenotaeniatus-A. spinifer assemblage also deserves attention. In Figure 1, A. stenotaeniatus and A. spinifer are placed within a strongly supported subclade associated with A. beijiangensis, as indicated by high support values on the tree. This result does not by itself revise the taxonomic status of A. stenotaeniatus, but it indicates that the relationship between A. longipinnis, A. stenotaeniatus, A. spinifer, and A. beijiangensis should be reexamined using both morphology and nuclear evidence.
The two A. paradoxus accessions were not recovered as a monophyletic group. A. paradoxus_1 was placed with A. wuyiensis, whereas A. paradoxus_2 was placed near A. jishouensis. This pattern could indicate cryptic diversity within A. paradoxus, historical introgression, mitochondrial capture, or misidentification of one or both accessions. The taxonomic complexity of A. paradoxus is not unexpected because A. formosanus, A. fasciatus, and A. labiatus have previously been treated as synonyms of A. paradoxus in Taiwan.11 Future work should include voucher-based morphological reexamination of the two A. paradoxus samples, broader geographic sampling, and nuclear-marker validation.
The placement of A. hemispinus within A. parallens is consistent with earlier molecular concerns regarding the taxonomic status of A. hemispinus.12 Nevertheless, this result should be interpreted cautiously because Zacco platypus, which has been treated as a synonym of A. parallens in morphological comparisons,10 was not included in the present dataset. The monophyly of A. monticola and A. yunnanensis was recovered with strong support, suggesting that the sampled mitogenomes for these species are internally consistent.
This work also compares the molecular units with morphology-based species concepts. Some MOTUs correspond closely to currently recognized species, such as A. monticola and A. yunnanensis, as well as to the sampled Onychostoma taxa. Other MOTUs combine more than one nominal species, such as the A. longipinnis-A. iridescens-A. barbodon and A. hemispinus-A. parallens units, suggesting possible synonymy, recent divergence, or mitochondrial introgression. Still other patterns, especially the two separate placements of A. paradoxus accessions, suggest potential cryptic diversity or misidentification. This explicit comparison strengthens the link between molecular delimitation and traditional taxonomy.
Morphological taxonomy should not be dismissed; rather, it should be integrated with molecular evidence. Controversies in Acrossocheilus morphology arise because diagnostic characters can be plastic or ontogenetically variable. In particular, body coloration, lateral stripes, vertical bars, dorsal-fin serrations, mouth and lip structures, and scale-count characters may vary with age, sex, preservation, or geographic population. These factors can generate synonyms, homonyms, and misidentifications when small samples or non-voucher sequences are used. The 15 MOTUs should therefore be treated as candidates for integrative testing against voucher morphology, morphometrics, meristics, geography, ecology, and nuclear genetic data.
The relationship between Acrossocheilus and Onychostoma remains unresolved. In the present mitochondrial tree, the Onychostoma clade (C3) was nested between two Acrossocheilus clades, and C1 was more closely related to C3 than to C2. A similar lack of reciprocal monophyly has been suggested in broader cyprinine phylogenies.28 However, only three Onychostoma taxa were included here, and the conclusion is based on mitochondrial protein-coding genes only. Therefore, the present study should not be used alone to redefine generic limits. Broader sampling of Onychostoma and related genera, preferably with nuclear genomic data, is necessary to test generic monophyly robustly.
A major limitation of this study is its reliance on mitochondrial loci. Mitochondrial genomes are maternally inherited as a single linked locus and can be affected by incomplete lineage sorting, introgression, hybridization, selection, sex-biased dispersal, and mitochondrial capture. These processes can cause mitochondrial trees to differ from species trees and can mislead species delimitation. In addition, several recognized Acrossocheilus species and many populations are missing from the available mitogenomic dataset, and the three-species sampling of Onychostoma is insufficient for strong conclusions about generic boundaries. For these reasons, the proposed MOTUs are best interpreted as mitochondrial hypotheses that require validation with unlinked nuclear markers, genome-wide SNPs, denser geographic sampling, and voucher-based morphology.
Taken together, the phylogenetic, delimitation, and taxonomic results provide a coherent framework for future revision of Acrossocheilus. The mitochondrial data identify several stable units, reveal candidate cases of synonymy or misidentification, and highlight lineages where current taxonomy may not match matrilineal history. The strongest practical contribution of this study is therefore not a definitive taxonomic rearrangement but a prioritized list of taxa and species complexes that should be examined using integrative taxonomy.
Conclusion
Using 31 complete mitochondrial genomes and the concatenated 13 mitochondrial protein-coding genes, we reconstructed phylogenetic relationships and assessed species boundaries in sampled Acrossocheilus and related taxa. The analyses supported 15 candidate MOTUs under the most congruent bPTP and ABGD settings, revealed several taxonomically important complexes, and showed that reciprocal monophyly between Acrossocheilus and Onychostoma was not recovered in this mitochondrial dataset. These conclusions are deliberately moderated because mitochondrial evidence alone cannot definitively resolve species limits or generic boundaries. Future studies should add nuclear genes or genome-wide markers, include the missing Acrossocheilus species and additional Onychostoma taxa, examine voucher specimens and diagnostic morphology, and integrate geographic and ecological information. Such integrative work will be essential for a stable taxonomy of Acrossocheilus and for conservation and management of these freshwater fishes.
Acknowledgements
The authors would like to thank TopEdit (www.topeditsci.com) for linguistic assistance during the preparation of this manuscript.
Author Contributions
Conceptualization: YuHe Yang (Equal), TingTing Lin (Equal). Data curation: YuHe Yang (Lead). Formal Analysis: YuHe Yang (Equal), TingTing Lin (Equal). Writing – original draft: YuHe Yang (Lead). Supervision: TingTing Lin (Lead). Writing – review & editing: TingTing Lin (Lead).
Conflicts of Interest
The authors declare no competing interests.
Data Availability
All sequences analyzed in this study are publicly available from the National Center for Biotechnology Information (NCBI), and the corresponding accession numbers are provided in Table 1. The final concatenated alignment and analysis input files should be deposited as supplementary materials or in an appropriate public repository upon acceptance.
Ethics Statement
This study was based exclusively on publicly available mitochondrial genome sequences downloaded from NCBI. No live animals were collected, handled, or sacrificed, and no experiments involving humans or animals were conducted. The use of publicly available sequence data complied with relevant ethical and database-use guidelines; therefore, additional ethical approval was not required.