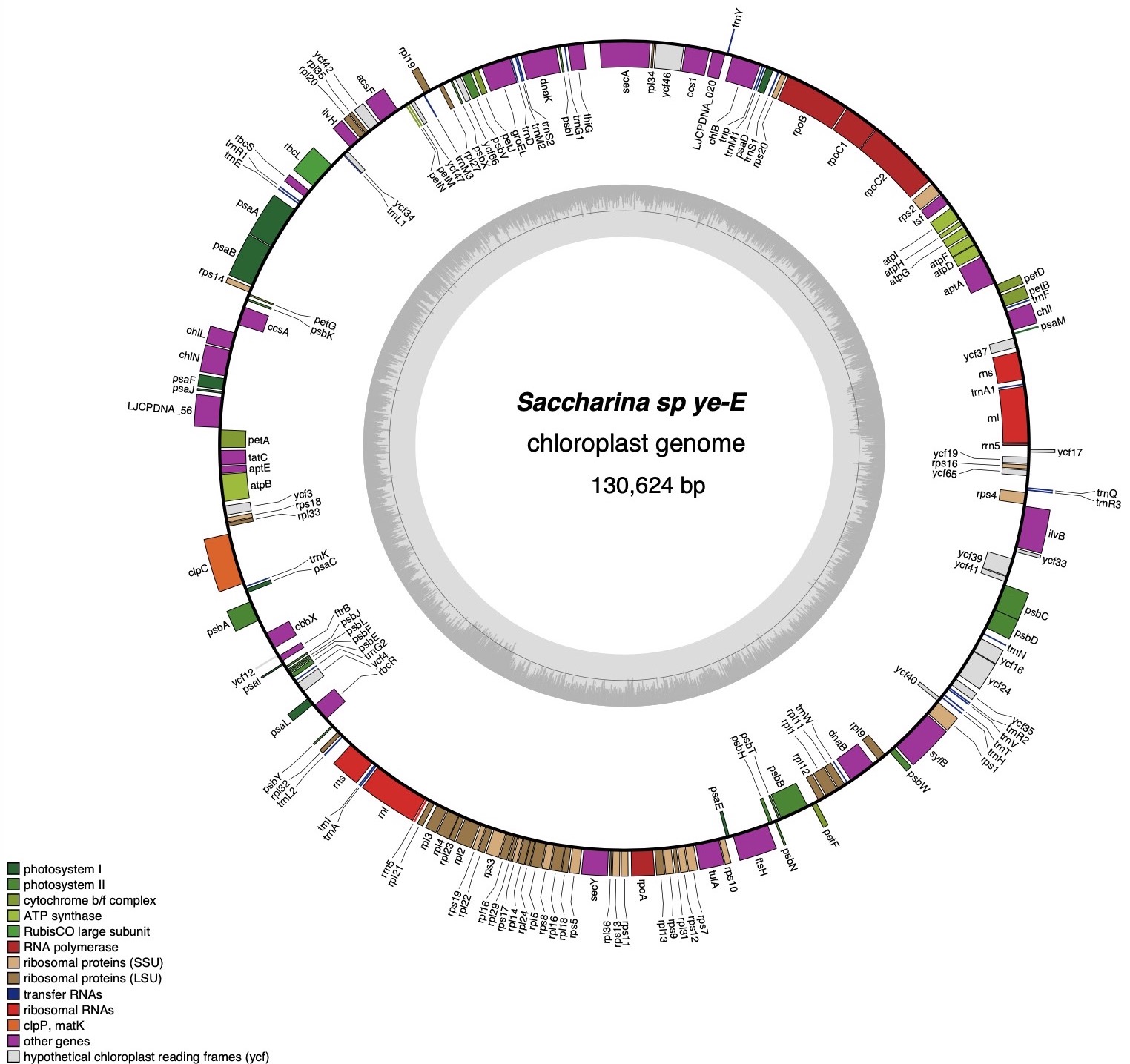

_in_the_chloroplast_genome_of_saccha.tiff)

Introduction
Saccharina, commonly known as kelp, is a genus of large brown algae in the family Laminariaceae. Saccharina species are native to many parts of the world, including the North Pacific, North Atlantic, and Southern Ocean. In addition to providing a habitat for various marine organisms, Saccharina species are an essential source of food for many marine organisms and have a range of ecological and economic uses. From an ecological standpoint, Saccharina plays a vital role in forming kelp forests, which absorb carbon dioxide through photosynthesis and provide a habitat for fish and other animals. From an economic standpoint, Saccharina is a major cultivated seaweed species, used not only as food but also as the primary raw material for extracting alginate, fucoidan, and mannitol. It is also highly valued in China, Japan, and Korea due to its commercial use as a raw material in food and industry.1,2
The degradation of Saccharina kelp forests is one of the most pressing threats to coastal ecosystems. Loss and decline of kelp forests have been reported in temperate, boreal, and arctic regions. They are mainly attributed to anthropogenic drivers such as climate change, eutrophication, overfishing, and habitat destruction.3 To understand and protect these critical ecosystems, phylogenetic research is urgently needed to identify genetic diversity among species, define evolutionary lineages, and elucidate the group’s evolutionary history. Such research can help us understand the evolutionary origins of this species, its current state of decline, and its potential for future recovery. With increased knowledge of the phylogeny of kelp species, it may be possible to develop conservation strategies to protect them in their current locations and restore populations in areas where they have been over-harvested or otherwise impacted. Additionally, phylogenetic studies can inform our understanding of how kelp species will respond to climate change and other environmental pressures in the future.4 Chloroplast DNA (cpDNA) is the most widely used marker for phylogenetic studies due to its high mutation rate, fast evolutionary rate, and maternal inheritance. cpDNA is a unique form of genetic material that is found in the chloroplastid of all photosynthetic organisms. As a result, cpDNA has been used in genetic studies to trace maternal ancestry and study species’ population structure. In plants, cpDNA studies have been used to understand phylogeny and evolution and assess genetic diversity in specified.5
The application of cpDNA in the genus Saccharina has been particularly useful for understanding the population structure of the species.6 Using cpDNA markers, researchers have identified different genetic lineages within the genus, which can help inform conservation and management decisions. For example, cpDNA analysis has revealed that different populations of Saccharina japonica have distinct evolutionary histories and that the species is composed of two distinct genetic lineages.6 This finding has significant implications for the conservation and management of the species, as different populations may require different management strategies. We analyzed the structural characteristics, simple repeat sequence (SSR) loci, relative species differences, codon preference and phylogenetic relationships in this study to determine the complete chloroplast genome sequence of a wild strain of Saccharina collected from Russia to increase genetic variance and economic value of japonica.
Materials and Methods
The Saccharina sp. ye-E sample used in the experiment was collected from the Sakhalin Oblast, Russia (50° 49’N, 142° 17’E). It was identified as Saccharina latissimi and labeled ye-E by the Yellow Sea Fisheries Research Institute of the Chinese Academy of Fishery Sciences. After the sample collection, it was cleaned with sterile water, dried with absorbent paper, frozen in liquid nitrogen and kept in an ultra-low temperature refrigerator for later use.
Obtain 0.1 g of Saccharina sp. ye-E samples and place them in a mortar that has been cooled using liquid nitrogen. After crushing, the genome was extracted using the CTAB method. Assess the purity and concentration of the DNA through 1% agarose gel electrophoresis and a multifunctional enzyme marker, and then submit it to a biological company for sequencing.
The paired-end reads were generated by using the Illumina sequencing system (Tianjin, China). Using Illumina sequencing, 152 million clean PE 100 reads of this DNA were obtained. The sequences were de novo assembled to contigs using ABySS Version2.07 firstly and then linked by referring to the chloroplast genome S. japonica (NC018523, 130,584 in length) using BWA (Burrows-Wheeler Aligner). Of the total reads, 32% were mapped to the reference chloroplast genome S. japonica. No gaps were present in the mapping result. The consensus sequence was produced with Geneious version 11.1.4 (http://www.genious.com). Additionally, the chloroplast genome was annotated by GeSeq,8 AGORA,9 CPGAVAS,10 OGDRAW11 MITOS server,12 the tRNAscan-SE server13 and PGA,14 which was also verified manually. Finally, the complete chloroplast genome of Saccharina sp. ye-E was submitted to GenBank (accession NO. MZ706293.1).
Employ the MISA (http://pgrc.ipk-gatersleben.de/misa/misa.html) software to search for Simple Sequence Repeats (SSRs), with the following parameters: a mononucleotide repetition threshold of 10, and a dinucleotide repetition threshold of 6; for trinucleotides, four nucleotides, five nucleotides and six nucleotides, the repetition threshold is 5. Utilize EMBOSS Explorer software (https://www.bioinformatics.nl/emboss-explorer/) to analyze the Effective Number of Codon (ENC), Relative Synonymous Codon Usage (RSCU), Codon Adaptation Index (CAI) and other pertinent parameters, where the RSCU value should be above 1 as the cut-off point, indicating that the codon is employed at a relatively high frequency; the ENC ranges from 21 to 60, and a value of 40 is the cut-off point for judging randomness in codons, while a value of 45 is the cut-off value for differentiating bias in codons. CAI values range from 0 to 1, with size proportional to codon preference.
Eight related species’ chloroplast genome sequences were downloaded from NCBI database, namely Laminaria rodriguezii (MT732096.1), Laminaria solidungula (NC 044690.1), Lessonia flavicans (MN561187.1), Macrocystis integrifolia (MW899036.1), Saccharina japonica (NC 018523.1), Saccharina sp. ye-B (MW038824.1), Saccharina latissima (NC 049039.1), and Undaria pinnatifida (KP298002.1), with Scytosiphon lomentaria (MK798154.1) and Ishige okamurae (MW762687.1) as the outgroup. The multiple sequence alignments of the selected chloroplast genomes were built using MAFFT software, and the phylogenetic tree was constructed by applying Neighbor-Joining, Mrbayes and Maximum likelihood optimality criteria using the concatenated nucleotide sequences shared among Saccharina sp. ye-E and the other 10 brown algae complete chloroplast genomes using IQtree2.15 All nodes were well supported by Bayesian posterior probabilities and 1000 bootstrap replicates.
Results
The total length of the chloroplast genome sequence of Saccharina sp. ye-E is 130,624 bp (Figure 1). It consists of 139 protein-coding genes (PCGs), 3 ribosomal (rRNAs) genes (5S rRNA, 16S rRNA, 23S rRNA, two copies for each) and 57 transfer RNA (tRNAs) genes. These 139 functional genes can be divided into three categories: 39 related to photosynthesis, 71 involved in genetic replication systems, and 29 other genes. The total GC content of Saccharina sp. ye-E is 31.1%. The nucleotide frequency of the H-strand is as follows: T, 34.5%; A, 34.4%; C, 15.4%; and G, 15.7%. The chloroplast of Saccharina sp. ye-E encodes 32,407 amino acids with the start codons included. The two 5S rRNAs are 110 bp and 100 bp, respectively; the 16S rRNAs are both 1,480 bp; and the 23S rRNA genes are 2,944 bp and 2,946 bp in length, respectively. Chloroplast data supporting this study are openly available in GenBank at the nucleotide database, Associated Bio Project (PRJNA272647), BioSample (SAMN03740579), and SRA (SRS947870).
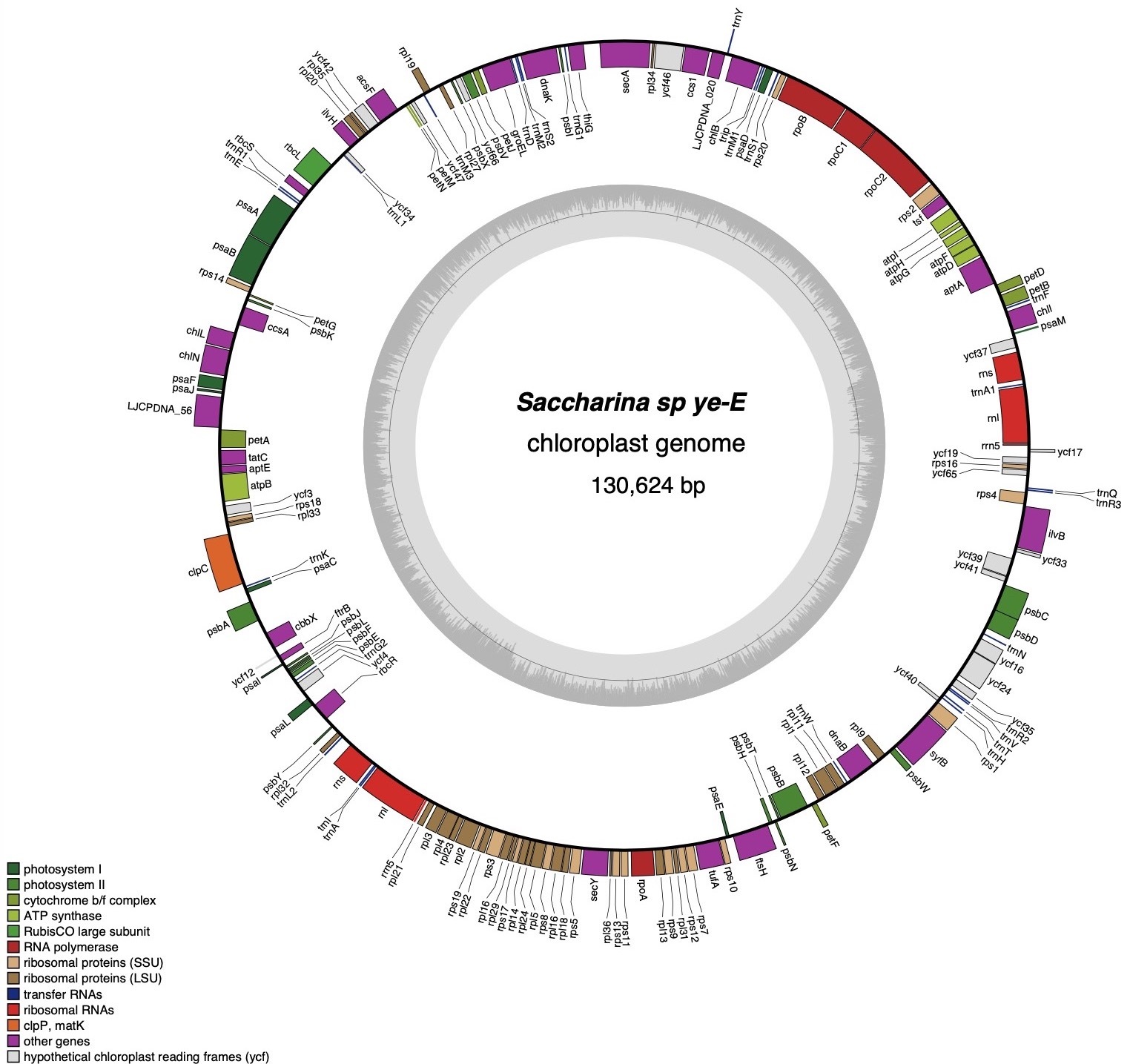
The chloroplast genome of Saccharina sp. ye-E contained 15 SSR markers, including three single nucleotides (A/T), which comprised 80% of the total. Additionally, two dinucleotides (AT/TA) and three trinucleotides (ATA) comprised 13.33% and 6.67% of the total, with no four, five, or six nucleotides being present in SSR (Table 1).
mVISTA analysis was used to compare the chloroplast genomes of Saccharina sp. ye-E with those of eight related species and two distant species(Outgroup), and it was observed that the related species shared the same structural composition, as shown in Figure 1, and Figure 2. The chloroplast genome of Saccharina sp. ye-E was found to be the largest and Macrocystis integrifolia had the smallest genome size, with a difference of around 1,300bp. The genetic composition and sequence of these chloroplast genomes were found to be highly conserved, except the Ishige okamurae and Scytosiphon lomentaria.

The use of codon bias can be employed to assess the compromise between codon divergence and natural selection in the process of translation. The EMBOSS Explorer analysis revealed that Saccharina sp.ye-E’s chloroplast genome had an effective codon number (ENC) of 51.666 and a codon adaptation index (CAI) of 0.617, which suggested that there was a powerful codon inclination in the chloroplast (Figure 3).
_in_the_chloroplast_genome_of_saccha.tiff)
The phylogenetic analysis showed that Saccharina sp. ye-E was resolved in a clade with S. latissimi (Figure 4). Support values based on Neighbor-Joining/Mrbayes/Maximum likelihood methods were strong enough to maintained a sister relationship in Saccharina genus.

Discussion
The characteristics of the chloroplast genome of Saccharina sp. ye-E were consistent with the typical features of angiosperm chloroplast genomes. SSR, or microsatellite, is defined as short sequence repeats of 1-6 bases which are found in the chloroplast genome of most plants. The amount of SSR in the chloroplast genome is lower than that in mitochondrial genomes, however it is inherited by one parent in the chloroplast genome, so it can still be used for species identification and population genetics studies.16 The presence of SSR in the chloroplast genome is essential for its molecular identification and resource conservation. Dissimilar to other reported chloroplast genomes, no four-nucleotide, five-nucleotide, or six-nucleotide SSRs were identified in the chloroplast genome of Saccharina sp. ye-E. Among all genes, rRNA was found to be the most conserved, which is consistent with previous research.17 Furthermore, codon preference is affected by genome size, gene length, gene expression level, and gene density. To guarantee the accuracy and dependability of the analysis results, three methods (Neighbor-Joining, Mrbayes, Maximum likelihood) were chosen to construct the phylogenetic tree. These results concur with the phylogeny obtained from the mitochondrial genome.18 The identification of the chloroplast genome of Saccharina sp. ye-E will be beneficial for future algal genetics evolution and systematic studies in the Laminariaceae.
Acknowledgments
This work was financially supported by Laoshan Laboratory (LSKJ202203204); the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (grant number 2021QNLM050103-1); the National Natural Science Foundation of China (grant number 32000404, 41606038, 41976110, 42176035).
Authors’ contribution
All authors contributed to the study’s conception and design. Wei Zhang and Ziwen Liu performed material preparation, data collection, and analysis. Wei Zhang wrote the first draft of the manuscript, and all authors commented on previous versions. All authors read and approved the final manuscript.
Conceptualization: Xiao Fan, Wei Zhang, Ziwen Liu;
Methodology: Wei Zhang, Ziwen Liu;
Writing - original draft preparation: Wei Zhang,
Writing – review, and editing: Xiao Fan, Wei Zhang, Ziwen Liu;
Resources: Xiao Fan.