Introduction
Streptococcus agalactiae, or group B streptococcus (GBS), is an important Gram-positive pathogen responsible for morbidity and mortality in fish aquaculture worldwide.1–3 GBS is also an emerging pathogen associated with neonatal meningitis in humans, mastitis in cows, and hematosepsis in rabbits.2,4,5,5,6 GBS can be classified into ten serotypes (Ia, Ib, and II to IX). The serotypes Ia, Ib, and III are regarded as the most predominant in fish GBS infection.7,8 In fish farms, transmission of GBS is possible through direct contact among individuals with cohabitation or indirectly through immersion in contaminated water of culture systems.9
Whole-genome sequencing (WGS) technology has become a fast and affordable tool that assists research in genetics, microbiology, ecology, and public health surveillance and response.10 WGS captures the full extent of bacterial genomic diversification and allows for genome-wide comparisons of clinical isolates with each other.11 Comparative genome analysis between bacterial strains, which are greatly different in host specificity or virulence, may help to rapidly screen for dispensable genes, gene deletions or mutations, and differentially-expressed proteins.12 Thus, WGS is an effective way of studying the mechanisms of cross-host infection, immunogenicity, pathogenicity, and resistance genotypes of bacteria. At present, hundreds of complete genomes of GBS have been sequenced. However, our understanding of the piscine GBS at the whole genome level is currently limited. Although some complete genome sequences and draft genome sequences of piscine GBS isolates have been recorded so far,9,13–17 the correlation between WGS analysis and phenotypic characteristics has not been reported. Here, this is the first report on draft genomes of GBS S03 and S07 isolated from Schizothorax spp. with different pathogenicity and drug resistance. Draft genomes of GBS S03 and S07 are important additions to the GenBank database of piscine GBS.
In the current study, antimicrobial susceptibility testing, experimental infection, and WGS analysis were carried out to compare two GBS clinical strains isolated from Schizothorax spp. Phylogenetic analysis was performed to identify their evolutionary relationships in GBS. We attempted to assess the use of WGS to evaluate the genomic diversity and genotypic prediction of drug-resistant phenotype and pathogenicity of them.
Materials and Methods
Bacterial strains
GBS S03 and S07 were isolated from diseased Schizothorax spp., with characteristic clinical and pathogenic meningoencephalitis. These strains were preserved in the College of Veterinary Medicine, Sichuan Agricultural University, China. Serotyping assigned GBS S03 to serotype III and GBS S07 to serotype Ia by cps cluster as described previously.18
Antimicrobial susceptibility tests
Antimicrobial susceptibility to vancomycin (VAN), penicillin (PEN), cephalexin (CFX), enrofloxacin (ENR), ofloxacin (OFX), norfloxacin (NOR), tetracycline (TET), doxycycline (DOX), erythromycin (ERY), gentamicin (GEN), florfenicol (FLO) and clindamycin (CLIN) was performed by the disc diffusion method. Antimicrobial susceptibility test results were interpreted according to Clinical and Laboratory Standards Institute (CLSI) breakpoints.19 Escherichia coli (ATCC 25922) was used as the reference strain for quality control according to CLSI guidelines. The results were collected from three repeated independent tests.
Pathogenicity experiments
To confirm the pathogenicity and virulence of GBS S03 and S07, we injected the two strain suspensions into healthy Schizothorax spp. and Danio rerio, intraperitoneally. Healthy Schizothorax spp. (11.5 ± 1.2 g) and Danio rerio (0.4 ± 0.05 g) were purchased from Sichuan Ya-fish Company and Model Animal Research Center of Nanjing University, respectively. 260 Schizothorax and Danio rerio were kept in 13 groups equally, respectively, in 120-L plastic tanks (n = 20/tank or group) with aeration and were acclimatized at 24 ± 1°C for 7 days. The Schizothorax of 6 experiment groups was challenged with 0.1 ml of bacterial suspensions of the GBS S03 and the other 6 groups were injected with the same volume of GBS S07 suspensions in sterile 0.8% NaCl solution at concentrations of 1.0×104, 1.0×105, 1.0×106, 1.0×107, 1.0×108, 1.0×109 cfu·ml-1. The control fish (n = 20) were injected with 0.1 ml 0.8% NaCl solution. Similarly, the Danio rerio of test groups were injected intraperitoneally with 0.05 mL of bacterial suspensions of the GBS S03 and S07 in sterile 0.8% NaCl solution at the abovementioned concentrations. The control animals were injected with 0.05 mL 0.8% NaCl solution. Mortality was recorded daily for 21 d and mean lethal dose (LD50) values were calculated. All the fish were euthanized by using 300 mg·L-1 MS222 (yuanye Bio. Co. Ltd., Shanghai, China).
Whole genome sequencing and annotation
To explore further differences between GBS S03 and S07, their draft genomes were sequenced and analyzed at whole genome level. The genomic DNA of the two strains were extracted using the TIANamp bacteria DNA kit (Tiangen, Beijing, China) and applied for library preparation using the Nextera DNA sample preparation kit (Illumina, San Diego, USA). The samples were accessed for the genome sequencing on an Illumina HiSeqTM 2000 (Illumina Inc., San Diego, USA) using a paired-end 2×100 bp protocol. The whole genome sequences were assembled using CLC Genomics Workbench 10.0 software (QIAGEN, Hilden, Germany). Contigs were initially annotated using Rapid Annotations using Subsystems Technology (RAST) and then manually checked. Resistance-related genes of the two strains were analyzed using the ResFinder 2.1 server.20 Gene clusters were classified into COG (Cluster of Orthologous Groups of proteins) categories (http://www.ncbi nlm.nih.gov/COG/) according to BLAST annotations.
Identification of antibiotic resistance genes and virulence factors
To find antibiotic-resistant genes in two GBS strains genomes, known antibiotic resistance genes were downloaded from the Comprehensive Antibiotic Resistance Database (CARD) and aligned with all coding sequences (CDSs) of those genomes using BLAST (https://card.mcmaster.ca/). A similar method has been applied to identify virulence genes from the Virulence Factor Database (VFDB) (http://www.mgc.ac.cn/VFs/).
Phylogenetic tree
A phylogenetic tree of GBS strains was constructed based on seven housekeeping genes (adhP, pheS, atr, glnA, sdhA, glcK, and tkt) as previously described.21 The alignments of these genes were concatenated into a single sequence alignment and a maximum likelihood tree was reconstructed using MEGA (version 10.0). The allele number and sequence type (ST) were assigned by the multilocus sequence typing (MLST) website (https://pubmlst.org/organisms/streptococcus-agalactiae).
Results
General features of the GBS S03 and S07 genomes
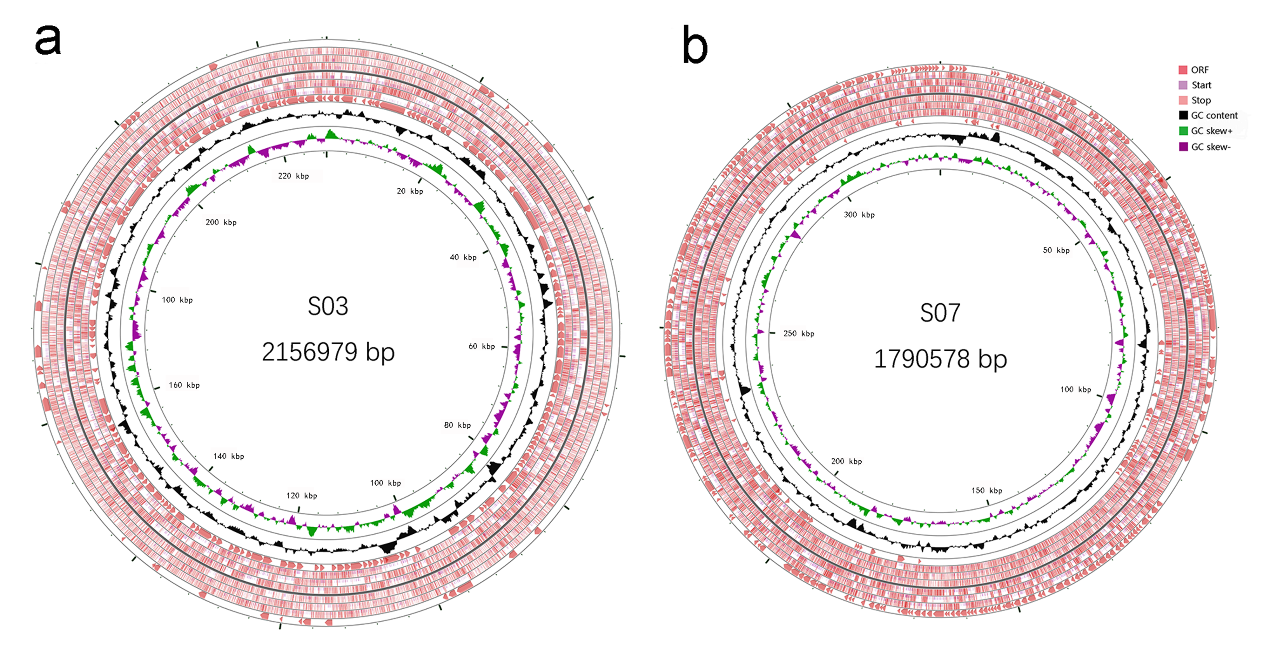
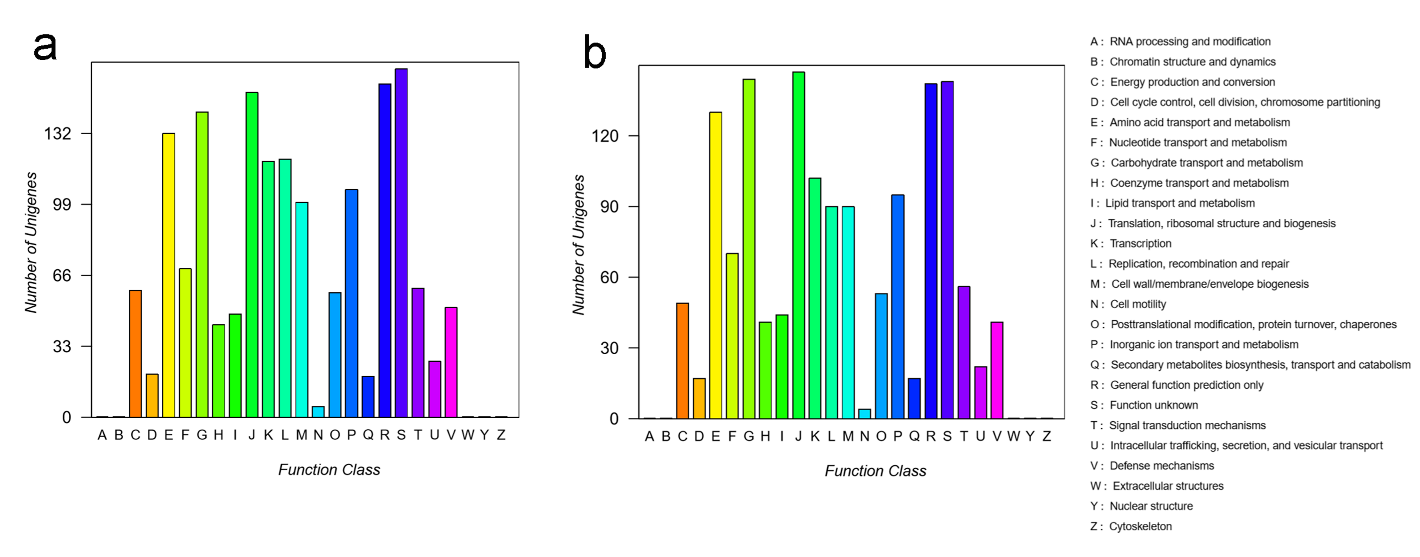
Characteristics of the two genomes are summarized in Table 1. The circular maps of the two strains showed that the genome of GBS S03 was 2,156,979 bp in length (Figure 1a), while that of GBS S07 was 1,790,578 bp (Figure 1b). The genome sequence of both strains showed 35% G+C contents. RAST server predicted the 2,160 and 1,641 CDSs for GBS S03 and S07 strains, respectively. Based on the COG classification of GBS S03 and S07, 23.7% and 22.6% of CDSs in GBS S03 were responsible for information storage and processing, whereas 37.5% and 39.4% of CDSs in GBS S07 were for metabolism (Figure 2). The genome sequences of GBS S03 and S07 have been deposited in the NCBI GenBank database under the accession numbers NAPX00000000 and NHZY00000000, respectively.
_and_gbs_s07_(b)_genome__generated_using_cgview_(version_2.png)

Antibiotic resistance profile and antibiotics resistance genes
Results of three repeated antimicrobial susceptibility tests for GBS S03 and S07 are given in Table 2. Antimicrobial tests revealed that GBS S03 showed drug resistance against macrolide, fluoroquinolone and tetracycline. In this study, a set of 12 antimicrobials was employed, and it was shown that S03 exhibited resistance to 7 of these antimicrobials, resulting in a resistance rate of 58.3%. By contrast, GBS S07 was sensitive to 11 tested antibiotics except enrofloxacin (intermediate), the resistance rate in this study was 0. The results indicated that the drug resistance of GBS S03 was stronger than that of S07 and had multiple drug resistance.
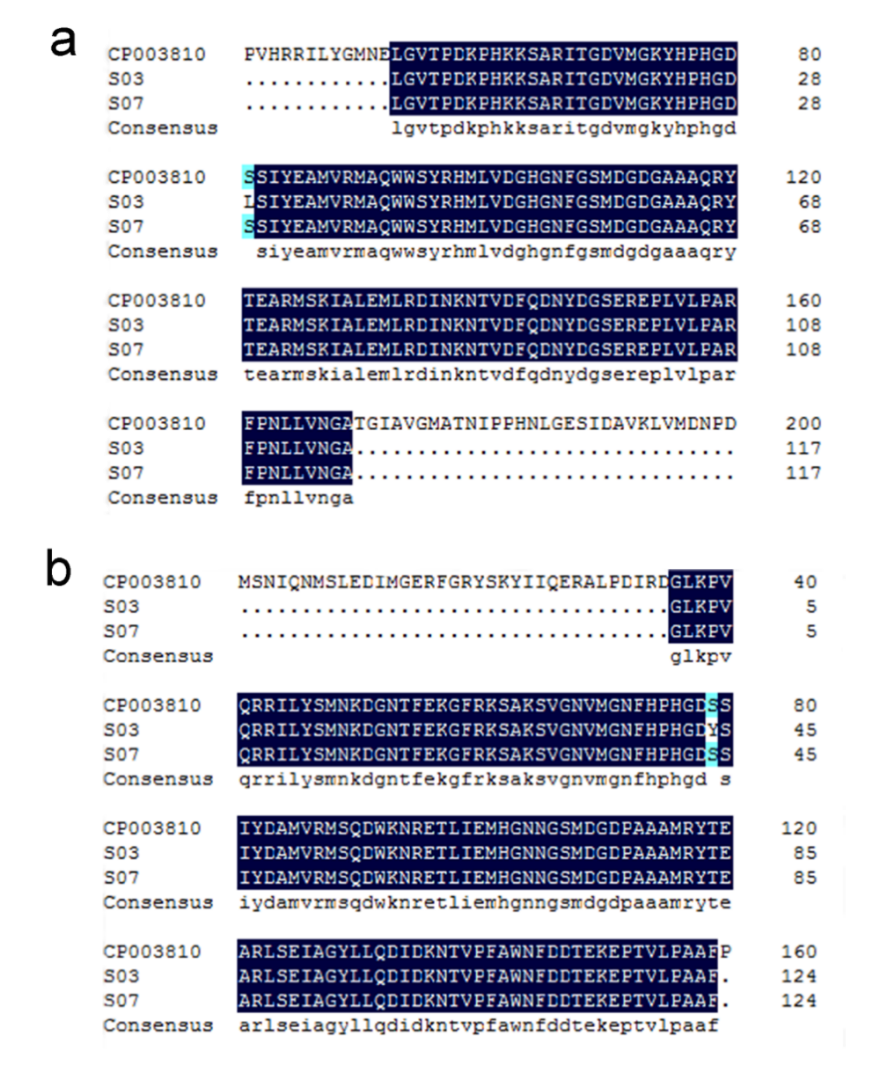
The genomes of GBS S03 and S07 showed that GBS S03 had 20 resistance genes, while GBS S07 had 14 resistance genes (Table 3). In addition to two macrolide resistant genes (macB and ermB) and one tetracycline resistant gene tetM, GBS S03 also had mefE and tetO genes. Meanwhile, GBS S03 had additionally lnuB, lsaE, APH3’, and sat-4 genes not present in GBS S07. Both strains carried genes encoding resistance to fluoroquinolone: parC, gyrA, gyrB, norA and norB, however, just GBS S03 had chromosomal gyrA [81:S-L] and parC [79:S-Y] mutations (Figure 3).
_and_*parc*_(b)_from_gbs_s03_a.png)
Pathogenicity experiments and virulence genes
At 21 days post-infection (dpi), GBS S07 induced higher levels of mortality and revealed a greater virulence than GBS S03 in the pathogenicity experiments. The mortality rates of infected fish are shown in Table 4. Concretely, the LD50 value of GBS S03 for Schizothorax spp. and Danio rerio was 5.27×108 cfu·ml-1 and 7.09×107 cfu·ml-1 respectively, while that of GBS S07 was 8.9×103 cfu·ml-1 for Schizothorax spp. and 1.88×103 cfu·ml-1 for Danio rerio. There was no mortality in the control groups during the observation period.
Fifty-one virulence genes were identified in both genomes (Table 5). Among these, bibA, dltA, fbsA, pavA, psaA, srtA, fbsB, pavB, lap, lep, lmb and srr-1 were involved in attachment; CLL_A 2400, hylB, lgt, mf3, and sugC were involved in invasion; cfb, cpsA-L, cpsA-Y, manA, neuA-D, uppS, rgpA, rmlA, scpA, rgpB, scpB, oppF, rgpG, sip, BC5263, EFD32, EFD32_0765 and SMU .322c were involved in evading/destroying host defenses; and other virulence genes, included were coxK2, clpC, clpE, clpP, cppA, msbA, nanA, gbpB, fhuC, licD, lisR, lplA1, groEL, htrA/deg, CT396 and stp. Comparison of the virulence genes between GBS S03 and S07 showed differences. GBS S03 carried SAG1404-1408, rib, SAK_0778-0779 and gbs0628-0629 associated with attachment genes, while GBS S07 were carrying SAN_1519, rfbA and cylE involved in invasion genes.
Phylogenetic analysis
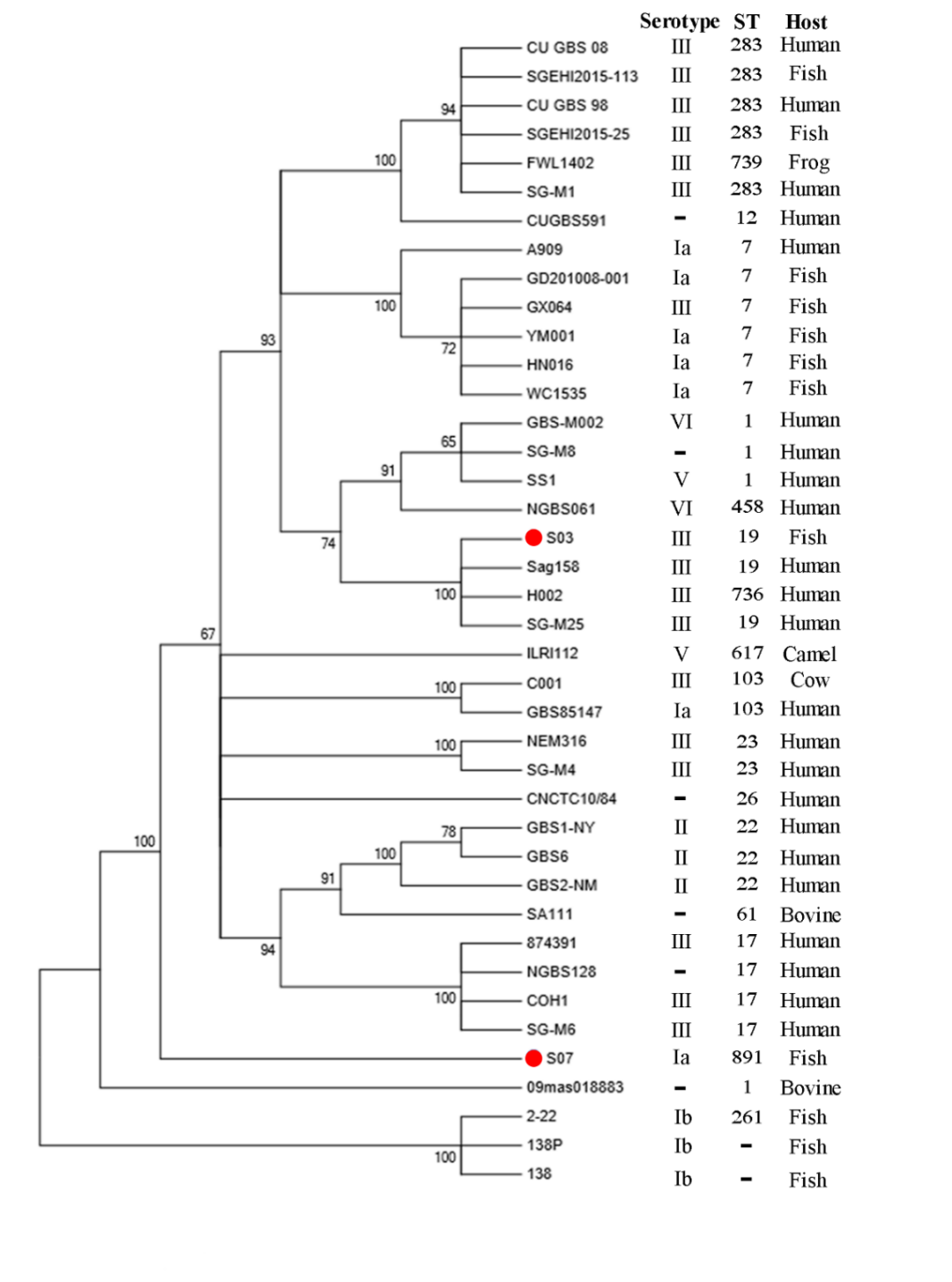
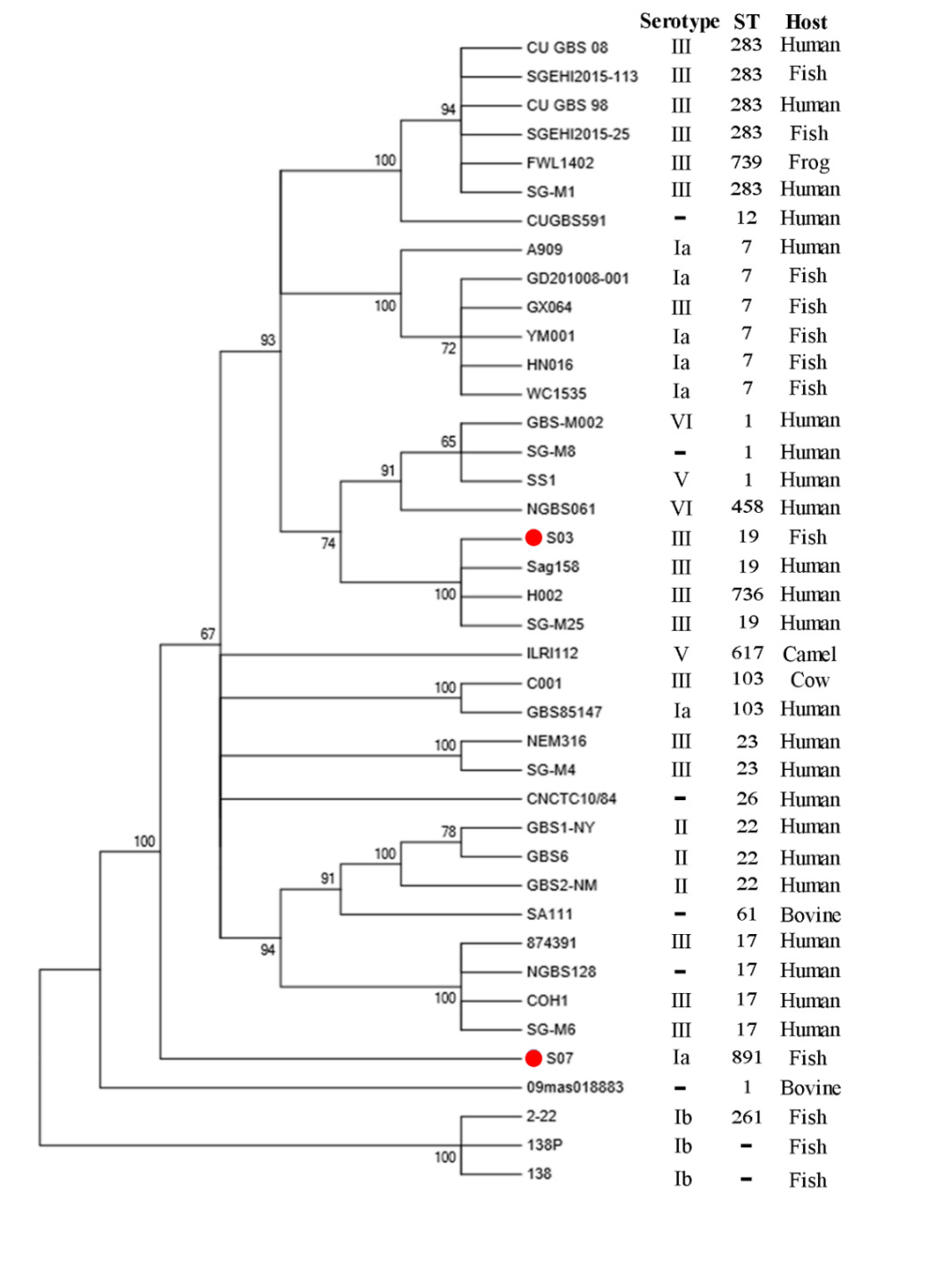
By MLST, GBS S03 was determined to be ST19, while GBS S07 was ST891. To investigate the phylogenetic relationships of our two isolates to other GBS strains, we used the maximum likelihood method to construct a phylogenetic tree (Figure 4). Phylogenetic analysis was performed using 38 reported GBS strains. Interestingly, GBS S03 was closely related to Sag158, H002 and SG-M25 strains isolated from human while GBS S07 was grouped separately.
Discussion
Since 2008, GBS has been present and emerging in tilapia in China. Zhang et al.22 and Liu et al.14 reported the first complete genome sequence of the piscine GBS strain isolated from cultured tilapia in China. In this study, draft genomes of GBS S03 and S07 isolated from Schizothorax spp. have been recorded. The genome size of GBS S07 was approximately 1.79 Mbp and approximately 0.37 Mbp smaller than GBS S03. There were more predicted CDSs in the GBS S03 genome than in GBS S07. Genome size is the result of a balance between amplification and loss of DNA, which evolves toward an optimal state for organismal fitness.23,24 Furthermore, genomic changes leading to streamlined genomes with increased pathogenicity and virulence are important.25 They might involve fragment recombination and contribute to the different virulence of GBS S03 and S07.
Antimicrobial tests revealed that the GBS S03, showed multidrug resistance, but GBS S07, showed drug-sensitive to eleven antibiotics except fluoroquinolones. In contrast to GBS S07, GBS S03 exhibited a stronger resistance towards macrolides, fluoroquinolones, and tetracycline antibiotics. In comparison to the WGS results of the two strains, it was shown that GBS S03 exhibited a greater abundance and complexity of resistance genes, especially those associated with drug resistance of macrolides, lincosamides, tetracyclines and aminoglycosides. The phenotypes of the two strains were in concordance with genomic characteristics revealed by WGS. Fluoroquinolones exert antibacterial activity by inhibiting DNA gyrase (DNA gyrase) and Topoisomerase IV (Topo IV). DNA helicase is composed of gyrA and gyrB subunits, and DNA topoisomerase IV is composed of parC and parE subunits.26 Two strains had fluoroquinolone genes: parC, gyrA, gyrB, norA and norB, however, just GBS S03 had chromosomal gyrA [81:S-L] and parC [79:S-Y] mutations, changing the target structure of fluoroquinolones in GBS. Therefore, GBS S03 has a high level of fluoroquinolone resistance.27–29 Current investigations have proven that there are two mechanisms of macrolide drug resistance: ermB encoding ribosome methylase modifies the binding site, mefA and mefE can change the active drug efflux system by changing the encoding efflux pump protein. In addition to two macrolide resistant genes (macB and ermB), GBS S03 additionally had mefE, which contributes to high-level macrolide resistance.28 The phenomenon of tetracycline resistance is attributed to the encoding of ribosome protection proteins, which effectively hinder the binding of ribosomes.30 The genes tetM and tetO are associated with the synthesis of ribosome-protective proteins. The tetracycline resistance of GBS S03 is notably enhanced compared to GBS S07 due to the presence of the tetO gene in addition to tetM.28,31 Meanwhile, GBS S03 had additional lnuB, lsaE, APH3’, and sat-4 genes not present in GBS S07 strain, the first two of which play an important regulatory role in lincosamide resistance.32 The gene lnuB is responsible for encoding a nucleotide transferase enzyme that causes clindamycin resistance.33 lsaE is a multi-drug resistance gene that can encode ABC transporter and mediate drug resistance of three drugs with different chemical structures: linkeramines, truncated laterin and streptomycin A.34 The combination of APH3’ and sat-4, associated with transposon Tn5405 among Gram-positive bacteria, encodes resistance to aminoglycosides (except gentamicin) and streptothricins.35,36 These results reveal a trend towards concordance between resistance phenotype and genotypic evidence for antibiotic resistance.
The pathogenicity of the two strains was compared by intraperitoneal injection in Schizothorax spp. or Danio rerio, which demonstrated that GBS S07 showed much greater virulence than GBS S03. GBS S07 also carried more virulence genes associated with invasion, such as SAN_1519, rfbA and cylE genes. However, it was counter to the previous report on GBS from tilapia in Thailand,16 which is most likely related to the diversity in host, area and strain. Fifty-one virulence genes in both GBS S03 and S07 involved in attachment, persistence, evading host defenses, tissue penetration, and toxin-mediated diseases had been identified in the GBS genomes, providing evidence of the pathogenicity of GBS.37–39 Differences in virulence genes between the two strains are mainly reflected in the genes encoding pili, including SAG_1404-1408, gbs0628-0629 and SAN_1519. Genes encoding pili in GBS are located within two distinct loci, denoted pilus islands 1 and 2 (PI-1 and PI-2), and comparative analysis of available genomes revealed two variants of PI-2, designated PI-2a and PI-2b.40 Gbs0628-0629 gene, SAG_1404-1408 gene, and SAN_1519 gene are located in PI-1, PI-2a and PI-2b, respectively.40,41 Pili are supposed to be important virulence factors.41 In GBS, pilus 1 is related to the immune escape, pilus 2a confers formation of biofilms, and pilus 2b is linked to the ability of attachment and invasion.42,43 In this study, GBS S03 carried PI-1 plus PI-2a type pilus islands while GBS S07 carried PI-2b type pilus island, which suggested there was concordance between pathogenicity and WGS result. Previous studies showed that most of the fish-sources strains carried PI-2a type, PI-2b type or PI-1 plus PI-2b type pilus islands, while in human strains, a great number of GBS carried PI-1 plus PI-2a type or PI-1 plus PI-2b type.44
In summary, based on genome-wide analysis combined with phenotypic experiments, we demonstrated a correlation between the two. The phenotypic findings of our study indicate that the GBS S03 strain exhibited greater resistance, whereas the GBS S07 strain had higher virulence. Correspondingly, in the results of whole genome sequencing, the drug resistance gene of GBS S03 strain was more complex. GBS S03 exhibited the presence of genes that govern resistance to multiple drugs. In order to prevent the future enhancement of GBS resistance mechanisms, it is imperative to minimize the overuse of antibiotics or explore other antibacterial ingredients(traditional Chinese medicine monomer) in practical aquaculture. The results of phenotypic testing indicate that GBS S07 exhibits greater virulence compared to GBS S03. The WGS result reveals a higher correlation between the pilus islands encoded by the virulence gene of GBS S07 and attachment and invasion, hence contributing to its greater virulence. However, whether our two strains in this study will exchange and transfer resistance genes and virulence genes in nature, with the appearance of new strains with great toxicity and extreme drug resistance, deserves further research and attention. Additionally, phylogenetic analysis showed that GBS S03 was closely related to human sources (Sag158, H002, and SG-M25) while GBS S07 was grouped separately. Combined with genome size and phenotypic characteristics, we hypothesized that they had different origins and evolutionary directions.
Further, bestowed pili types and phylogenetic analysis, we hypothesized that GBS S03 had a common source with human GBS or the possibility of cross-infection with human strains. Unfortunately, we do not have adequate epidemiological information to confirm this hypothesis. Although the knowledge about the ability of GBS to cross the interspecies barrier and allow human-derived strains to infect animals, or vice versa, is poorly understood,45 demonstrated that GBS strains from human and bovine origins could infect fish. Priyanka et al.46 found that fish-isolated GBS could infect humans by consuming raw fish. Therefore, we should attach great importance to cross-host transmission of GBS and its potential threat to public health security.
Acknowledgments
This work was supported by the Fish Diseases Prevention and Control Project of JPDC (No.23XB0027) and Sichuan Innovation Team Project of Agricultural Industry Technology System (SCCXTD-15)
Authors’ Contribution per CRediT
Writing – original draft: Yihao Wang (Lead). Validation: Yu Yuan (Lead). Formal Analysis: Kun Peng (Lead). Data curation: Yilin Wang (Lead). Project administration: Longjun Deng (Lead). Supervision: Tiancai Li (Lead). Investigation: Defang Chen (Lead). Conceptualization: Ping Ouyang (Lead). Funding acquisition: Xiaoli Huang (Lead). Resources: Hongrui Guo (Lead). Visualization: Huidan Deng (Lead). Methodology: Weiming Lai (Lead). Writing – review & editing: Yi Geng (Lead).