Introduction
The pen shell, A. pectinate, of the family Pinnidae, is a large wedge-shaped bivalve mollusc naturally distributed along the Indo-West Pacific coast from southeast Africa to Melanesia and New Zealand.1 As a benthic bivalve with a life span of up to 7 years, the pen shell, living in muddy to sandy substrates, plays an important role in maintaining the ecological stability of seagrass beds, lagoons and coral reefs1 The family Pinnidae in the Indo-Pacific. Indo-Pacific Moll. 1:175–226. It is also an economically important mollusc in many countries, including China, Korea and Japan. In China, as one of the Eight treasures of seafood, its large, nutritious adductor muscle is traditionally dried and preserved as ‘conpoy’. As a lucrative commercial fishery, A. pectinate has suffered continuous population declines due to over-fishing, habitat loss, pollution, and other factors.2 Therefore, knowledge of the population genetic structure of A. pectinate is essential for the sustainable management of natural resources. Using mtCOI and nrITS genes, Liu et al investigated the species diversity and evolutionary history of five A. pectinata from different geographical groups, supporting that these five morphological forms corresponded to five divergent genetic lineages.3 A genetic structure analysis of three A. pectinata populations along the west coast of South Korea was performed using 13 SSRs and found no significant genetic differentiation.2 The mitochondrial cytochrome c oxidase subunit I (COI) gene and seven microsatellite loci were used to assessed the population genetic structure of eight A. pectinate populations, including seven from northern coast of China and one from the north coast of Korea.4 The results showed that there was no genetic differentiation between China and North Korean intraspecific populations, and low but significant genetic differentiation between the Northern China and North Korean populations. These results provided important clues for the genetic conservation and management of A. pectinate along the Bohai Sea and Yellow Sea. However, the pen shell populations, especially those with variable shells, are mainly distributed in the South China Sea,5 and until now, the comparative analysis of genetic diversity and population structure of A. pectinate between the North China Sea (NCS) and South China Sea (SCS) has never been carried out.
To accurately analyze the level of genetic diversity and gene flow within and among populations of A. pectinata in the north and south east coast of China, genic SSR (expressed sequence tag SSR, EST-SSRs) were developed to analyze the genetic structure of A. pectinata collected from five natural habitats along the coast of Bohai Sea (BHS), Yellow Sea (YS), East China Sea (ECS) and South China Sea (SCS). Compared with genomic SSRs, genic SSRs are more conservative and have relatively higher transferability, especially in cryptic species.6 Meanwhile, with high divergence and hybridization, six pen shell populations along China coast have been revealed as cryptic species by molecular and morphological data,7 so the EST-SSRs are more suitable for the genetic structure analysis of A. pectinate. To date, EST-SSRs have been widely used to study the population genetic structure of barley (Hordeum vulgare), sugar beet (Beta vukgaris), Pacific white shrimp (Litopenaeus vannamei) and pearl oyster (Pinctada maxima).8–11 Although many EST-SSRs have been developed for A. pectinatethe identification and polymorphism validation of EST-SSRs has mainly been carried out.12 To our knowledge, no EST-SSRs have been used to analyse the population genetic structure of A. pectinate from different geographical locations along the Chinese coast.
In the present study, five populations of A. pectinate and its genus, Atrina vexillum, as an outgroup were collected from 6 different habitats along the Chinese coast. Nine polymorphic EST-SSR markers were used to assess the genetic diversity within and between populations and to estimate the level of gene flow between them, which will help in the conservation, genetic management, and utilization of A. pectinate.
Materials and methods
Sample collections and Genomic DNA preparation
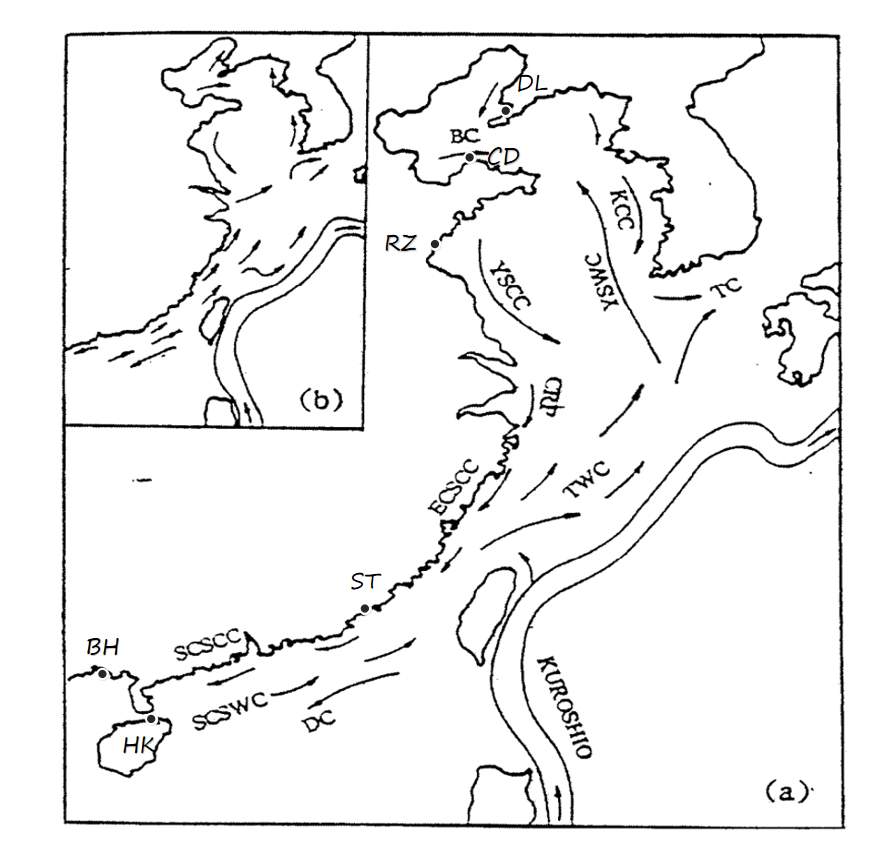
Five wild A. pectinate populations comprising 148 individuals were collected from five coastal locations in China between 2021 and 2022. Three populations were obtained from the North China Sea (NCS) including the Dalian population (DL), the Changdao population (CD) from the Bohai Sea (BHS) and the Rizhao population (RZ) from the Yellow Sea (YS); the other two populations were sampled in the South East China Sea (SECS), including Shantou (ST) and Haikou (HK). In addition, 36 individuals of Atrina vexillum from the Beihai Sea (BH) were included as an outgroup were included, giving a total of 184 samples (Table 1, Figure 1). The number of individuals in each population ranged from 28 to 36. Adductor muscle tissue was dissected from each sample and immediately preserved in liquid nitrogen, and then stored in a freezer at -80℃. Total genomic DNA was extracted from the adductor muscle using the TIANamp Marine Animals DNA Kit (Tiangen Biotech Co. Ltd., Beijing, China). DNA quality was assessed by agarose gel electrophoresis (1%) and quantity was determined using a Qubit fluorometer (Life Technologies), then the DNA was diluted to 20 ng/μl for further polymerase chain reactions (PCRs).
_winter__(b)_summer._local_currents_are_.png)
Microsatellite analysis
Ten EST-SSR markers with high levels of heterozygosity for A. pectinate were analyzed, including AP-28, AP-29, AP-31, AP-41, AP-43, AP-1511, AP-0245 (developed by our library), c55835, c67433 and c6902612 (Table 2). The forward primers of each pair were labeled with a fluorescent dye (6-FAM or HEX; Applied Biosystems, Foster City, CA, United States) at their 5’end. PCR was performed with 5μl Premix Taq (TaKaRa Taq version 2.0 plus dye), 0.1μl of each primer (10μmol/L), and 1μl template DNA in a final reaction volume of 10μl. Thermocycling conditions were as follows: 94℃ for 3 min, 30 cycles of 94℃ for 30 s, 50-60℃ for 20s, 72℃ for 3 min, and an extension at 72℃ for 5 min. PCR products were electrophoresed and genotyped on an ABI Prism 3730XL automated DNA sequencer (Applied Biosystems), and allele range was assessed using DNA sequencing analysis software v5.3.0 (Applied Biosystems) with a ROX 500 size standard.
Statistical analysis
The effect of null alleles, large allele dropout and allele scoring errors were checked using MICROCHECKER 2.2.3 software prior to population genetic analysis.14
The number of alleles (NA), effective alleles (NE), observed (HO) and expected (HE) heterozygosity, Hardy-Weinberg equilibrium (HWE) analysis, and inbreeding coefficient (Fis) were calculated using the GENALEX v6.5.15 To investigate genetic differentiation between and among populations, analysis of molecular variance (AMOVA) and estimation of pairwise F-statistics (FST) values were performed using Arlequin 3.5 with 10,000 permutations.16 Multiple simultaneous tests of FST values were adjusted using the sequential Bonferroni procedure.17 The effective number of migrants per generation (the gene flow, Nm) was estimated using the equation: Nm = (1-FST)/4FST. To standardize the number of alleles among different populations, a minimum of 26 samples per population were used to estimate allelic richness (Ar) using the program FSTAT v2.9.4.18 Genetic distances (Dc) were estimated using the chord genetic distance method,19 and a neighbor-joining tree (NJ tree) was constructed using the MEGA5.0 software based on Dc. The Mantel test was used to assess the correlation between genetic and geographic distance using GenAlEx6.5. Model-based Bayesian clustering in STRUCTURE version 2.2 was used to investigate the patterns of population structure further. The K-values were set between 1 and 3. For each value of K, the calculations were performed with a burn-in of 10,000 followed by 100 000 iterations under an admixture model with correlated allele frequencies. Three mutation models of IAM, TPM, and SMM with 1000 replications were used to analyze the evidence for a genetic bottleneck in each population using the Bottleneck1.2.02.20
Results
Variation of markers
According to the result of genotype analysis, these nine microsatellite loci were all highly polymorphic. A total of 174 alleles, including 115 and 59, were detected in five populations of A. pectinate and one outgroup of A. vexillum, respectively. The number of alleles (Na) per locus varied from 8 (JY-29) to 29 (c67433), and the average number of alleles at the locus level ranged from 4.667±0.333 (JY-41) to 18.833±1.327 (wc55853) (Table 3). The mean effective number of alleles (Ne) per locus in all samples ranged from 1.984±0.091 (JY-41) to 11.822±1.460 (wc55835), and the allelic richness (Ar) per locus ranged from 4.395±1.249 (JY-43) to 18.263±2.953 (wc55835). The mean allelic richness for each population was as follows: HK (8.255), CD (8.422), ST (8.556), DL (8.889), BH (9.921), and RZ (9.734). No significant difference in mean Ar was found between the six populations (P= 0.9995). The mean values of observed (Ho) and expected heterozygosity (He) ranged from 0.391±0.092 (JY-31) to 0.933±0.029 (JY43) and from 0.430±0.096 (JY-1511) to 0.908±0.012(wc55853), respectively. On average, the outgroup of the BH population had the lowest He value (0.620), and the DL population had the highest (0.691) (Table 3).
Linkage disequilibrium was assessed for all 216 pairwise combinations in 6 populations using Arlequin 3.5 software, the results showed that 25 pairwise linkage disequilibria were detected at the significant level p=0.05. The observed heterozygosity (Ho) were tested for concordance with the HWE (Table 3). Twelve cases of locus-population combinations (BH, DL and HK at c67433; CD at JY28; CD, DL, HK, RZ and ST at c5585; HK, RZ and ST at JY43;) out of 54 (six populations-nine loci) showed significant deviation from HWE (P<0.001, after sequential Bonferroni correction =0.05/54).17 Of these, eight out of 12 deviations showed significant heterozygote deficiencies (Ho<He). The possibility of null alleles was detected using MICRO-CHECKER; the results suggested the presence of null alleles at wc55835 in all populations and c67433 in five populations (except BH). The genotypes of wc55835 and c67433 were therefore excluded from further analysis.
Population Genetic Structure
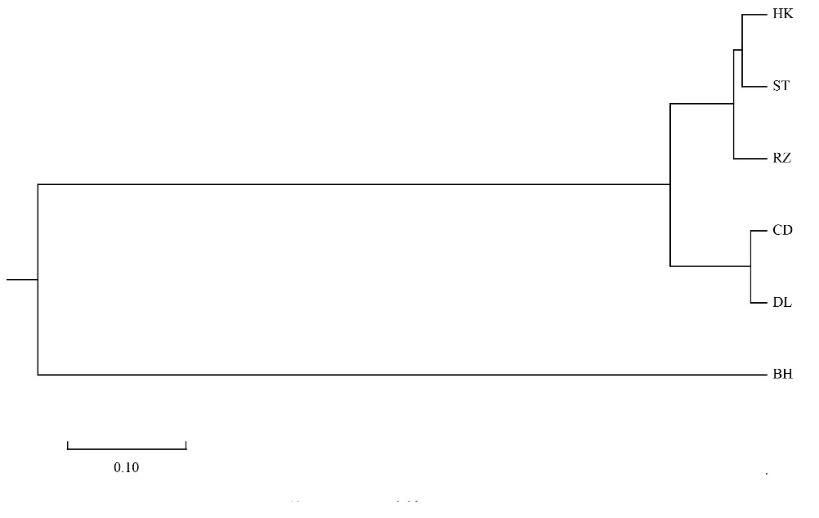
The pairwise genetic distance (Dc) and genetic identity (IN) are shown in Table 4. Among five populations of A. pectinate, the pairwise Dc varied from 0.027 (DL-CD) to 0.191 (CD-RZ), and the genetic identity varied from 0.826 to 0.974 (Table 4). A neighbor-joining tree was constructed using Nei’s genetic distance (DC) values between five A. pectinate populations and A. vexillum as an outgroup (BH) based on seven microsatellite loci. The five A. pectinate populations were divided into two clusters, with the CD and DL populations from the Bohai seas (BHS) forming the first group, followed by the HK and ST populations from South East China Sea (SCES), and then with the RZ population from the Yellow Sea (YS) forming the second group (Figure 2).

Although this clustering result was partly consistent with their geographical origin, the RZ population did not cluster with the northern populations (DL, CD), suggesting that there is no significant genetic divergence and geographical differentiation between the North China Sea (CD, DL and RZ) and the Southeast China Sea (HK and ST) (Figure 2). According to the NJ tree topologies, AMOVA analyses were performed on A. pectinate for the levels of genetic differentiation between populations among groups, among populations within groups, between individuals within populations (Table 5). The results showed that most of the variation was within populations (93.389%), followed by among groups (5.829%) and among populations within groups (0.782%), with among populations within groups and within populations showing significant variation (P<0.05).
Pairwise FST values ranged from 0.008 (between DL and CD) to 0.342 (between BH and HK) (Table 6). Eleven of the 15 pairwise FST values were significant after Bonferroni correction (P < 0.003, Bonferroni correction = 0.05/15), among which the pairwise FST values between A. vexillum (BH) and A. pectinate (DL, CD, RZ, HK and ST) showed relatively high genetic differentiation ranging from 0.302 (BH-DL) to 0.342 (BH-RZ), whereas the pairwise FST values between five A. pectinate populations were only 0.008 (DL-CD) to 0.042 (CD-HK, CD-RZ), indicating that A. vexillum is genetically distinct from A. pectinate (Table 6). Pairwise FST analysis indicated that only DL-CD, HK-ST, HK-RZ and RZ-ST showed no genetic differentiation (Table 6), consistent with the NJ tree clustering results (Figure 2). The effective number of migrants per generation (Nm) in A. pectinate ranged from 5.651 (CD-HK) to 30.450 (CD-DL), suggesting considerable gene exchange especially between DL and CD, HK and ST, HK and RZ, ST and RZ (Table 6). For the bottleneck tests, no significant heterozygote excess was detected under three mutation models of IAM, TPM and SMM (Table 7), consistent with the normal L-shaped distribution of allele frequencies in each population. The result indicated that six populations had not experienced a genetic bottleneck in the recent past.
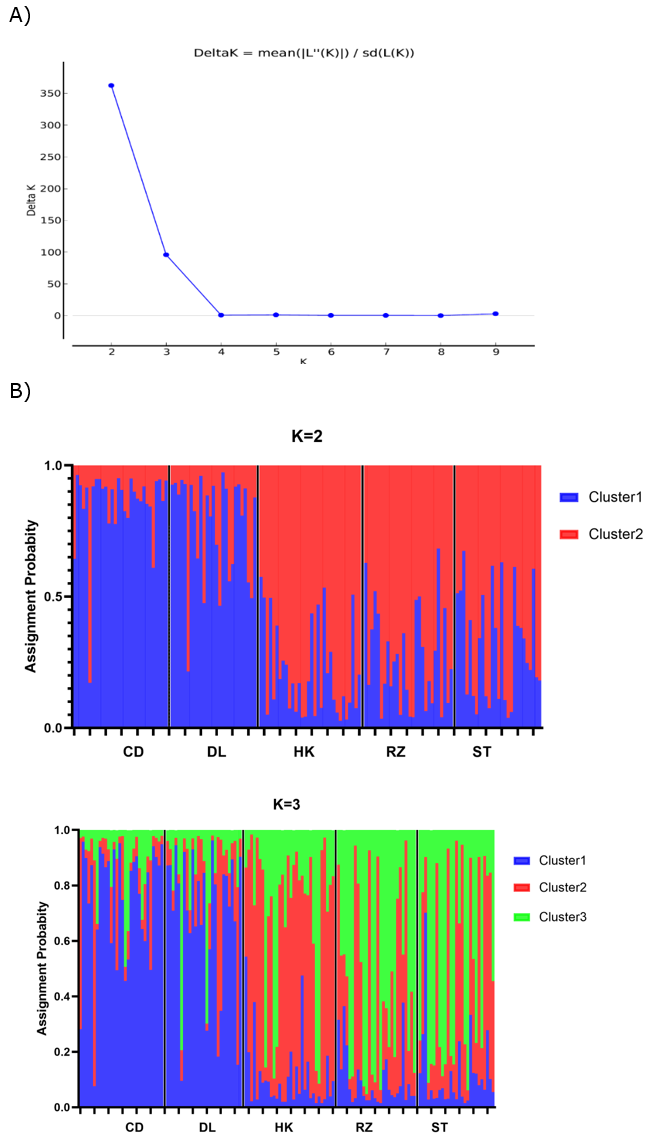
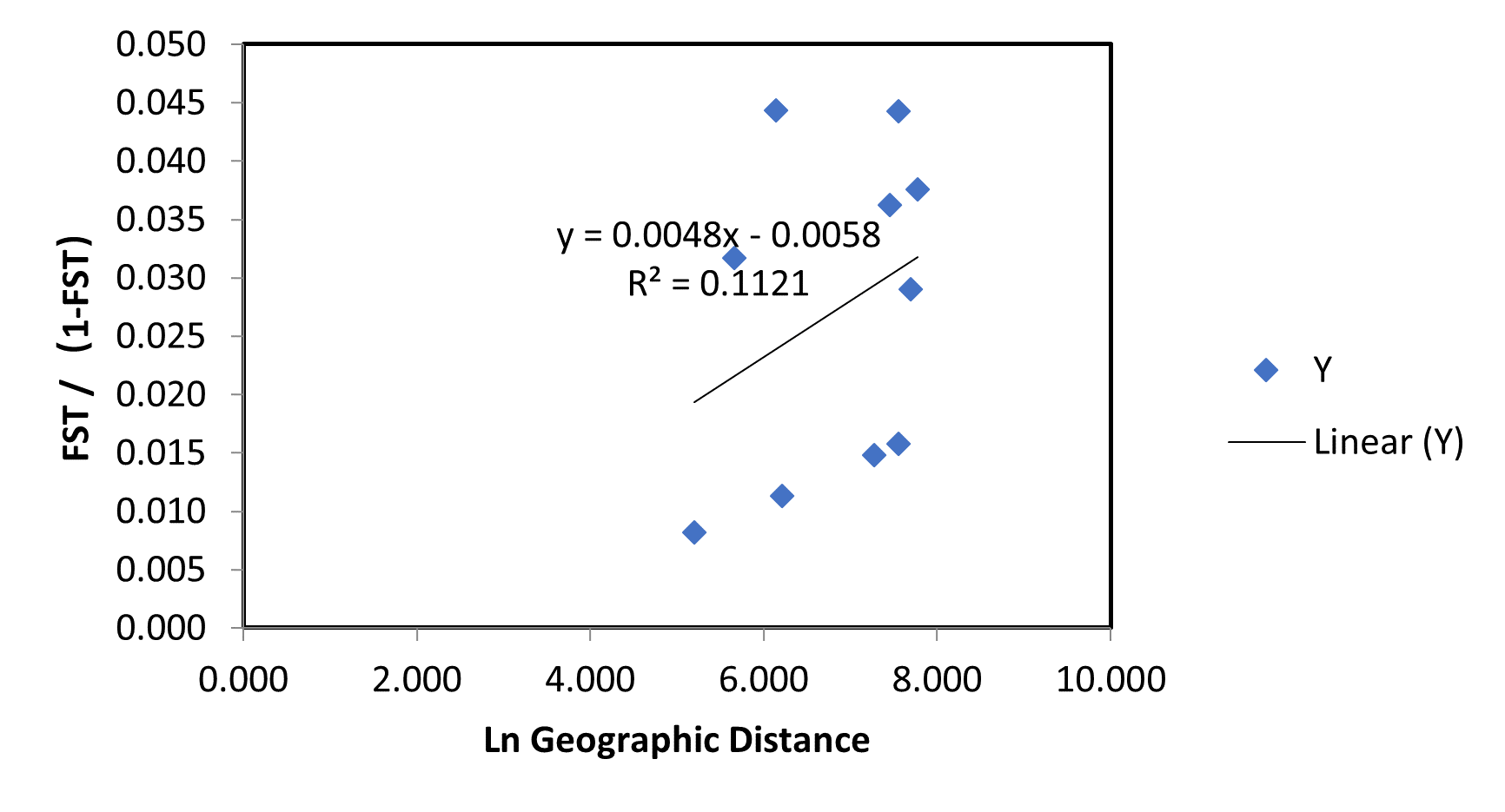
Genetic structure analyses used Structure 2.3 with the best-fit K=2 (Figure 3A). Although the results showed that the A. pectinate populations could be divided into two clusters, the five populations did not show any distinct structure (Figure 3B), suggesting that they were individual populations. Mantel test R-value for isolation by distance was 0.335 (P=0.150), indicating no significant relationship between genetic and geographic distance (P>0.05). This indicates no genetic divergence and geographic differentiation among A. pectinate populations along the Chinese coast (Figure 4).


Discussion
Genetic diversity and departures from HWE
Genetic diversity assessment is the basis for A. pectinate to establish breeding and conservation programs. In the present study, nine EST-SSR markers were used to study the genetic diversity of five A. pectinate populations including DL, CD, RZ, ST and HK, among which six EST-SSR markers were developed in our laboratory, the other three EST-SSR markers were identified by Sun et al.12 Based on these nine SSR markers, 115 alleles were detected in 148 samples of A. pectinate with mean values of Na=9.067, Ar=8.978, Ho=0.634 and He=0.679, respectively, which were higher than those detected in the EST-SSR development of A. pectinate by Sun et al.12 (with 3 out of 23 loci identical to those used in our study, i.e., c55835, c67433 and c69026). This result is due to the different genotyping method used by Sun et al.,12 which used silver staining to visualize genotypes. Compared to the sequencing-based genotyping used in this study, the resolution of the silver staining method is much lower than the sequencing-based genotyping, which significantly affected the allele detection rate. The genetic diversity in the present study was lower than that in Korea2,21and China of A. pectinate.4 The different SSR type may be the main cause of the low genetic diversity in our study. Some studies suggested that the average number of alleles for SSR primers derived from the non-coding DNA region (genomic SSRs) was higher than that of EST-SSR primers (genic SSRs).22,23 Similarly, the mean allele (Na) per locus of EST-SSR markers in this study was lower (8.911) than that of genomic SSR markers (16.03) in the genetic structure of A. pectinate by An et al.,21 which may be due to the location of EST-SSR markers in more conserved and expressed sequences compared to genomic SSR in relatively variable non-coding region, as shown in many studies.23–27
Most of the locus-population combination cases were in HWE, only 12 (BH, DL and HK at c67433; CD at JY28; CD, DL, HK, RZ and ST at c5585; HK, RZ and ST at JY43) of them showed significant deviation from HWE (p<0.05), and 8 out of these 12 cases (BH, DL and HK at c67433; CD, DL, HK, RZ and ST at c5585) showed significant heterozygote deficiencies (Ho<He), which was the main cause of deviation from HWE. Significant heterozygote deficiency has been frequently reported in marine invertebrates.2,4,28,29 Studies have suggested that null alleles are a potential cause of this phenomenon.28,30 In the present study, the null alleles at c55835 were detected in all six populations and c67433 in five populations (except BH), of which 8 were heterozygous deficient, consistent with the results of the HWE test (Table 2). This result indicates that null alleles play an important role in heterozygote deficiency and deviation from HWE, which was also found in Argopecten irradians,31 A. pectinate,4 Meretrix meretrix.28,29 The occurrence of null alleles is associated with mutations within the primer sequence that can prevent primers binding and can be avoided by primer redesigning.32 In addition, heterozygote excess was also detected at JY-43 in RZ, HK and ST (Ho>He).
4.2. Genetic structure and genetic differentiation
Knowledge of the population structure and gene flow is essential for effective management and conservation. In this study, the genetic relationships between the five populations were basically consistent with their natural geographic locations, except for RZ, which didn’t cluster together with the north populations (DL, CD) from the Bohai Sea but with the southern populations (HK, ST). Genetic differences in marine organisms between different geographical populations are often influenced by ocean currents,28,33 dispersal capacity,4 environmental tolerance and substrate and the interaction between these factors.34,35 It is well known that the Bohai Sea, a semi-enclosed sea, has a relatively low water exchange rate with the Yellow Sea,36,37 which to some extent hinders the migration of A. pectinate larvae from the Bohai Sea to the Yellow Sea, which may be the main reason for the separation of the RZ population from the CD and DL populations. Similar results have also been reported in other marine bivalves, including Scapharca broughtonii5 and Chlamys farreri.37
For marine organisms, the dispersal ability of larvae plays an important role in the formation of the genetic structure.38,39 Many marine organisms have pelagic larvae that can potentially link distant populations by dispersal on ocean currents.40,41 The planktonic larval duration (PLD) of A. pectinate is approximately 30 days with main reproductive season from April to September.4,42 In spring and summer, a warm, salty current known as the Taiwan Warm Current flows offshore of the Kuroshio, mainly through the Tsushima Strait into the Sea of Japan, but some of it flows intermittently north along the Korean coast into the Yellow Sea and merges with the Yellow Sea warm current (Figure 1).43 This means that for the long-lived planktonic larvae of the pen shell, the Taiwan Warm Current could transport the planktonic larvae of the ST population from the East China Sea to the Yellow Sea, although the migration may be affected by the summer Yangtze River outflow, which reduces the salinity,28,33 the wide range of salinity tolerated by A. pectinate may facilitate gene flow via long-distance dispersal of A. pectinate larvae.44
In addition, the geographical barriers caused by primary production, freshwater influence and turbidity are all present on continental margins and around high islands, which has implications for the effectiveness of barriers to larval dispersal.34,45 However, A. pectinate could live down to depths of 100m,4 so its larval dispersal might be less affected by the ecological or hydrographical barriers. Therefore, the larvae of A. pectinate from the ESC could overcome the barriers caused by the freshwater discharge of the Yangtze River and migrate to the Yellow Sea. These could be the reasons for the clustering of the RZ population from YS with ST from ESC and HK from SCS.
Despite the geographical barriers between the Yellow Sea and Bohai Sea, East China Sea and Yellow Sea, respectively, there was genetic exchange between the five populations of A. pectinate. According to this study, the pairwise FST values ranged from -0.003 to 0.059. Wright suggested that 0.05 < FST < 0.15 represented a moderate degree of genetic differentiation; whereas 0.15 < FST < 0.25 and FST > 0.25 indicate a high degree of genetic differentiation.42 A lower FST value indicates a higher level of gene flow and lower genetic differentiation between populations.22 Thus, in our study, low FST values were obtained within and between clusters (CD, DL, and RZ, ST, HK), indicating a high level of gene flow, and a relatively low genetic differentiation was detected among A. pectinate populations. The AMOVA test indicated no significant genetic differentiation within groups (p<0.05). In addition, the Mantel test (P>0.05) also suggested that the genetic structure of A. pectinate populations was not significantly influenced by geographical distances along the Chinese coasts (Figure 4). The same results were found in the population structure analyses of A. pectinate populations by An et al.,21 which show no genetic differentiation between populations from different locations, likely due to the high dispersal potential. Marine organisms with high dispersal potential generally show low levels of genetic differentiation over large geographical distances.3,46,47
Conclusion
The present study suggests that although the five populations were divided into two sub-clusters, strong gene flow exists between them, especially within each sub-cluster. No genetic divergence and geographic differentiation were found among A. pectinate populations. The high connectivity level and low genetic structure of A. pectinate along the Chinese coast were probably caused by gene flow through larval dispersal.
Acknowledgments
The study is supported by the Natural Science Foundation of Shandong Province (ZR2020MC192), the Agricultural Industrial Technology System in Shandong Province (SDIT-14), and the “First class Fishery Discipline” program in Shandong Province, China, a program in Shandong Province, China.
Authors’ Contribution
Writing – original draft: Peican Zhu (Equal), Fukai Wang (Equal). Writing – review & editing: Peican Zhu (Equal), Fukai Wang (Equal), Bo Liu (Equal). Conceptualization: Biao Wu (Equal), Chunde Wang (Equal). Resources: Biao Wu (Equal), Bo Liu (Equal). Formal Analysis: Feng Wang (Equal), Xiaotong Zhang (Equal), Kai Yu (Equal). Investigation: Feng Wang (Equal), Xiaotong Zhang (Equal), Kai Yu (Equal). Methodology: Chunde Wang (Equal), Bo Liu (Equal). Funding acquisition: Bo Liu (Lead). Data curation: Bo Liu (Equal). Validation: Bo Liu (Equal).
Peican Zhu and Fukai Wang contributed equally to this work
Conflicts of Interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical Approval
This article contains no studies with human participants or animals performed by authors.
Informed Consent Statement
All authors and institutions have confirmed this manuscript for publication.
Data Availability Statement
All are available upon reasonable request.