Introduction
Crabs are mostly inhabited in the intertidal zone of sea, estuary, rivers, and lakes. Crabs can adapt to changes in temperature and salinity in waters. To reduce cost, cultured mud crabs and Chinese mitten crabs were transported to market in an air-exposed environment. During transportation, crabs cannot effectively obtain enough oxygen and water, causing hypoxia stress. Hypoxia stress can hinder normal physiological metabolism and affect the physiological activity of crabs.1 Crabs must regulate many physiological and biochemical indexes to cope with air exposure stress.2,3 Long-term air exposure will disturb osmotic pressure and dry gills and destroy normal physiological metabolism.4 Finally, crabs will be dead. Therefore, hypoxia stress is an important factor affecting the economic benefits of the crab breeding industry. Understanding the molecular mechanism of air exposure stress in crabs is helpful for the transportation and culture of crabs.
In recent years, transcriptome analysis has been widely applied as a useful tool to screen candidate genes and pathways underlying molecular mechanisms.5 Transcriptome sequencing technology is characterized by low cost, high efficiency, and high throughput.5 It can reveal the genes and signaling pathways involved in air exposure stress at the transcriptome level and further elucidate its molecular mechanism.6,7
Mud crab Scylla paramamosain, which belongs to the family Portunidae and class Crustacea of the phylum Arthropoda, is widely distributed in the Indian-West Pacific region.8 In some countries such as China, Thailand, Vietnam, Indonesia, Philippines, and Malaysia, S. paramamosain has high commercial value in local and international markets.9,10 Soft-shell and sexually mature female crabs are high-end varieties of S. paramamosain.11,12 In 2018, the annual production of mud crabs worldwide was 415,500 tons.9 The production of mud crab in China was more than 15 hundred thousand tons in 2021.13 Cheng et al.14 analyzed the hepatopancreas transcriptome of S. paramamosain under air exposure and found that long-time air exposure would damage immune function and disrupt metabolism. Gills are the main respiratory tissues of crabs, which are exposed to many environmental factors. Nowadays, the research on gill transcriptome after air exposure was reported only in crab Eriocheir sinensis,6 Portunus trituberculatus,15 Corbicula fluminea7 and Cardisoma armatum.16 Several differentially expressed genes (DEGs) and signaling pathways related to immunity, osmosis, and substance and energy metabolism were screened and identified. However, there is no transcriptome analysis in the gill of mud crab under air exposure stress.
This study used RNA-seq to investigate the transcriptome response of gill in S. paramamosain after air exposure. The DEGs between the control and experimental groups were screened and enriched in GO and KEGG terms. The expression levels of six DEGs that play important roles in the air exposure process were examined by RT-PCR. This result will be useful to understand further the molecular mechanism under air exposure in mud crab and other crab.
Materials and Methods
Sample collection
Twelve healthy mud crabs of S. paramamosain (average body weight = 136 ± 5.3g) were purchased from a crab aquaculture farm in Nansha District, Guangzhou, Guangdong province. They were transported to the Zhuhai Base of the South China Sea Fisheries Research Institute (Zhuhai, Guangdong Province, China). All crabs were reared in a cement pool with a temperature of 25℃ and salinity of 8‰ and fed with clams and snails (Sinotaia quadrata). The seawater was renewed daily. After a week, eight healthy crabs were chosen randomly and divided into two groups. Four crabs in the experimental group (EG) were placed in tanks without seawater, and four crabs in the control group (CG) were reared in aerated seawater. After 8 h of exposure, gills from each individual were dissected and preserved in liquid nitrogen until used.
RNA extraction, cDNA library preparation, and transcriptome sequencing
According to the manufacturer’s instructions, total RNA was isolated from each sample using Tirzol Reagent (Invitrogen, USA). The quality of total RNA was examined by 1.0% agarose gel electrophoresis and a 2100-Bioanalyzer (Agilent Technologies, USA). Equal quantities of RNA from each group were pooled to construct a cDNA library, respectively. Libraries were generated using the NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA) according to the manufacturer’s introduction by the Shanghai OE Biotech Co., Ltd (Shanghai, China). After cluster generation, the library preparations were sequenced on an Illumina Hiseq 2500 platform, and paired-end reads were generated.
Quality control and de novo assembly
To obtain clean reads, the adaptor sequences, low-quality sequence reads (bases quality≦20), and reads containing poly-N larger than 35bp were removed from the raw reads by Trimmomatic software.17 The clean reads were assembled de novo with Trinity software.18 The TIGR Gene Indices Clustering tool removed redundant sequences and performed further assembly.19 All sequence reading segments were assembled from scratch to generate transcripts. Transcripts symbolized the significant parts of respective isoforms according to gene homology, and the longest transcripts of each gene were referred to as ‘unigenes’.
Gene functional annotation
All of unigenes were aligned with protein databases NCBI non-redundant protein sequences (NR), Conserved Domain Database (CDD), Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups of proteins (KOG), NCBI nucleotide sequences (NT), Gene Ontology (GO) and Protein family (PFAM) using an E value cut-off of 1 x 10-5 using diamond software.5 In addition, unigenes were aligned with the protein family database Pfam using HMMER.20 Gene names were assigned to each assembled sequence based on the best BLAST hit (highest score). To annotate the assembled sequences with GO terms describing biological processes, molecular functions, and cellular components, the Nr BLAST results were imported into Blast2GO.21 WEGO was used for GO functional classification of all unigenes.22 Additionally, pathway assignments were carried out based on the KEGG database.
Differentially expressed genes (DEGs) analysis
According to the screening criteria of DEG (q ≤ 0.05 and |log2FC|≥2), 20,128 DEGs were detected between CG and EG. The number of sequences reads that mapped to reference sequences was calculated using bowtie2,23 and the FPKM (Fragments Per Kilobase per Million) value, which represented unigene expression, was performed with eXpress to identify DEGs between the CG and EG.24 GO and KEGG enrichment analyses of DEGs were performed by topGO (version 2.24) and clusterProfiler (version 3.0.5), respectively.
Real-time quantitative PCR validation
Real-time quantitative PCR (RT-qPCR) was carried out to validate the result of RNA-seq data. Six DEGs, including anti-lipopolysaccharide factor 1(ALF1), crustin 6, crustin antimicrobial peptide (CAP), caspase-2, cystatin, and crustin 2, were chosen for validation. All primers used were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) (Table 1). The RNA samples used for qRT-PCR amplifications were the same as those used to construct the RNA-Seq library mentioned above. For each sample, quantification was performed using Prime-Script™ Reverse Transcriptase Reagent Kit with gDNA Eraser (TaKaRa, Japan). The β-actin of S. paramamosain was used as an internal reference.25 The qRT-PCR was performed in triplicate for each sample using the CFX96 real-time PCR Detection System (Eppendorf, Germany) in a 25 μL reaction system, with the following components: 12.5 μL of 2 × SYBR Green Real-time PCR Master Mix (Takara, Japan), 0.5 μl of each primer (10 μM), 1.0 μL of cDNA, and 10.5 μl of RNase-free water. The PCR program was as follows: 95°C for 30 s, 40 cycles of 95°C for 5 s, and 60°C for 30 s. A melting curve was obtained at the end of the reaction to assess the specificity of the PCR amplification, and a single peak was observed. Data were quantified using the 2-ΔΔCT method.26 The amplification efficiencies of the target and reference genes were verified and found to be approximately equal. Differences between means were considered significant at the 95% confidence level (P < 0.05). The results were expressed as means ± standard errors.
Results
De novo assembly of S. paramamosain transcriptome
In this study, the gill transcriptome of S. paramamosain was sequenced and analyzed. More than 101 million pairs were generated from two groups (CG and EG): 52,911,676 reads in EG and 48,167,508 reads in CG (Table 2). A total of 52,202,626 clean reads and 47,491,150 clean reads were generated in EG and CG, respectively. The GC content was ranged from 40% to 43%. Both Q30 of raw and clean data were more than 96%, indicating that high-quality sequencing data was obtained. The raw read data were submitted to the NCBI Sequence Read Archive (SRA) database with the following accession numbers: PRJNA978973.
All clean reads were assembled de novo into 145,873 transcripts, with average lengths of 885.05 bp and N50 lengths of 1,785 bp (Table 3). Moreover, 90,014 unigenes were assembled. The number of unigenes above 500 bp length was 26,073, accounting for 28.97%. In addition, 12,804 unigenes (14.22%) were greater than 1,000 bp. The assembly program produced many high-quality long sequences available for subsequent annotation analysis.
Functional annotation
All unigenes were annotated successfully in several databases, including Nr, Nt, COG, CDD, Pfam, GO, and KEGG (Table 4). 38,507 unigenes (42.78%) were annotated in one database, and 1,050 unigenes (1.17%) were annotated in all databases. There were 34,768 unigenes annotated successfully in the Nr database, which was the most annotated database, followed by KOG and GO. The least annotated database is Nt, with 5,744 unigenes.
Identification and analysis of differentially expressed genes
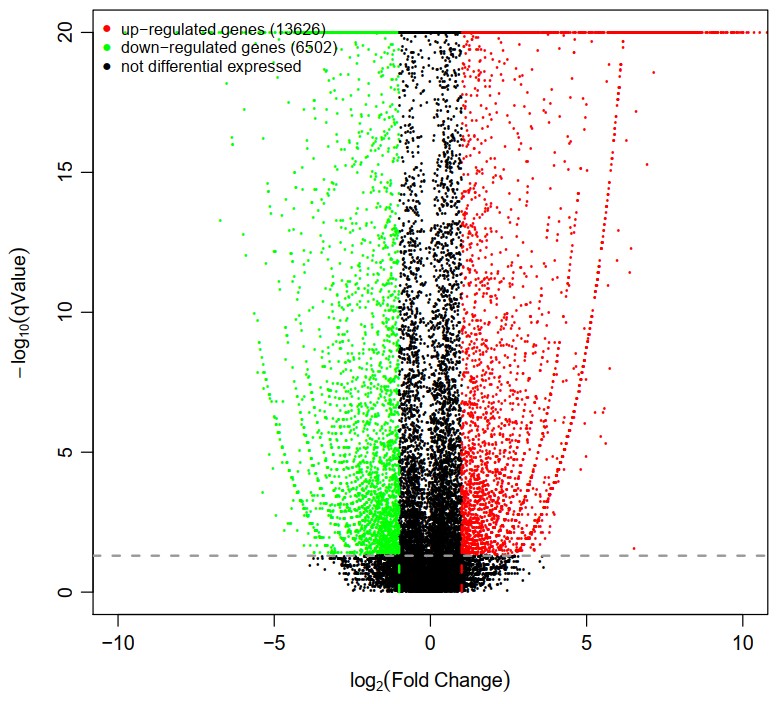
Compared with the CG, there were 13,626 up-regulated and 6,502 down-regulated DEGs (Figure 1). Some immunity-related genes were detected, for example, heat shock protein 70 (HSP70), HSP90, HSP22, selenium-dependent glutathione peroxidase, thioredoxin, C-type lectin, crustin 2, cathepsin L1, ALF1, glutathione S-transferase, Profilin, Cystatin, Cytochrome P450; Apoptosis related genes (Caspase-2) and metabolism-related genes (long-chain fatty acid coA ligase, apolipoprotein D, urea transporter 2, glutamine synthetase, lipase maturation factor, glucose dehydrogenase and glucose 6 phosphates) were obtained. Genes involved in ion exchange were found as follows: sodium/potassium/calcium exchanger (NCKX), Na+/K+ ATPase (NKA), ATPase Na+/K+ transporter (NKAT), and Na+/H+ exchanger (NHE).

DEGs enriched in GO and KEGG
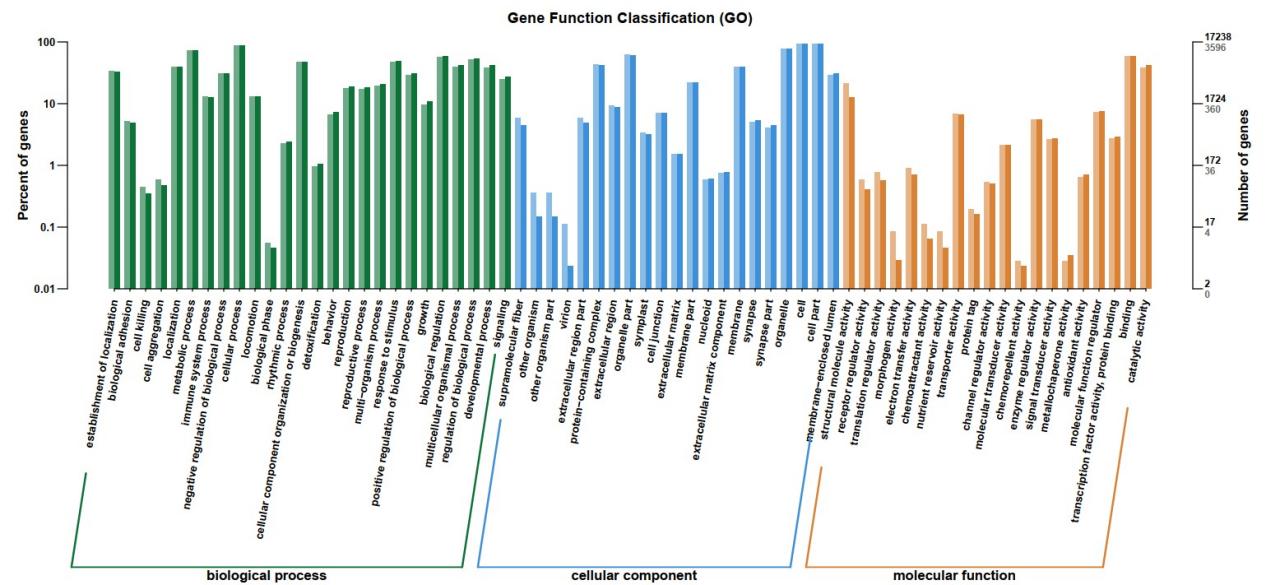
The analysis of DEGs enrichment in GO and KEGG were performed. According to GO functional analysis, DEGs were classified into three major GO terms: cellular component (CC), biological process (BP), and molecular function (MF). 3,332 DEGs were enriched into 26 categories of BP, and most DEGs were detected in subcategories of “metabolic process” (GO:0008152). 3,400 DEGs were found in 21 categories of CC, and the dominant subcategories under BP were “cell” (GO:0005623) and “cell part” (GO: 0044464). 3106 DEGs were enriched in 20 categories of MF, and the main subcategories were “binding” (GO:0005488) (Figure 2).

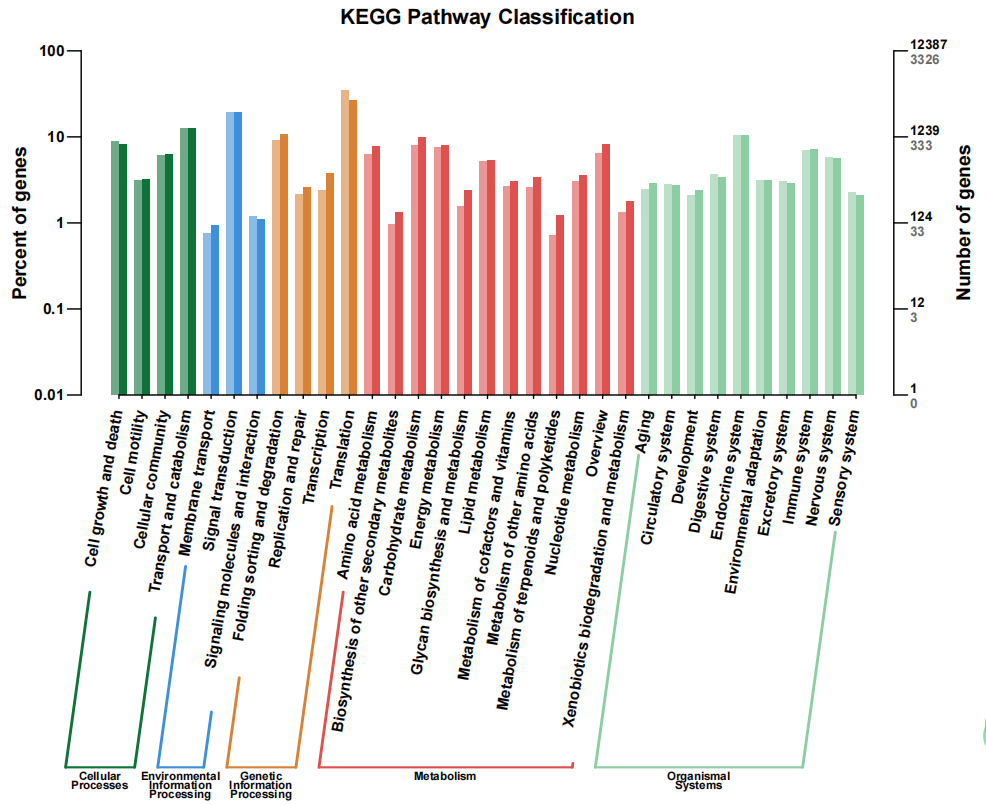
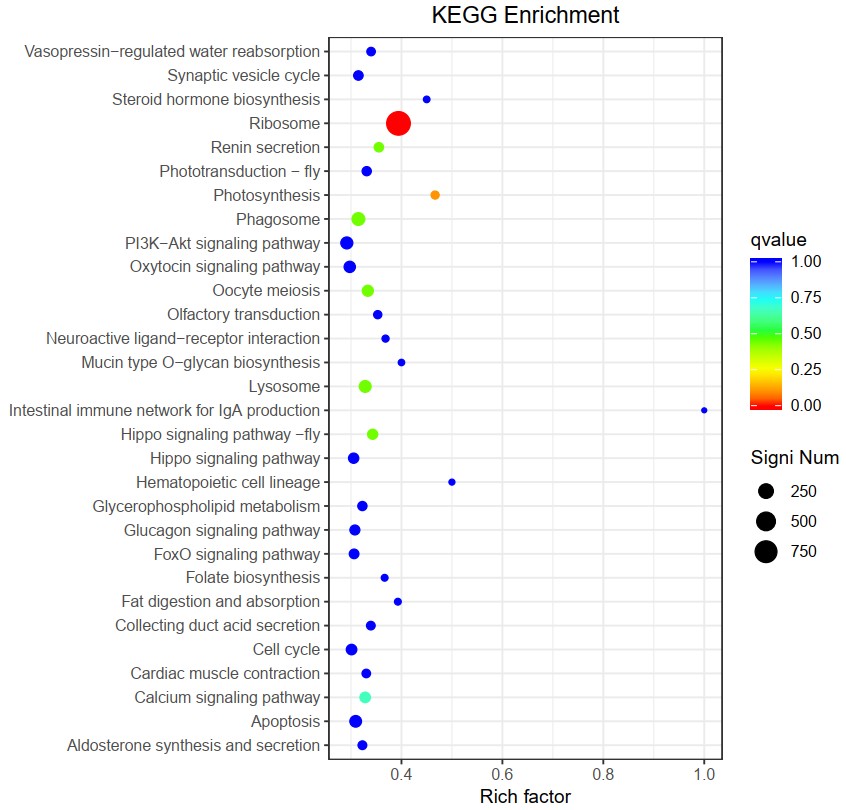
The KEGG pathway analysis showed that 3,326 DEGs were assigned to five KEGG terms: Cellular Processes, Environmental Information Processing, Genetic Information Processing, Metabolism, and Organismal Systems (Figure 3). The most enrichment term was observed in Genetic Information Processing (1,593 DEGs), followed by metabolism and Organismal Systems (1,531 DEGs). In addition, DEGs were distributed in 280 KEGG pathways. The top 30 most enriched KEGG pathways include some key functional categories, such as immunity (Lysosome, Hippo signaling pathway) and metabolism (Ribosome, Mucin type O-glycan biosynthesis, Glucagon, Glucagon signaling pathway, Folate biosynthesis, Calcium signaling pathway, Steroid hormone biosynthesis) (Figure 4).


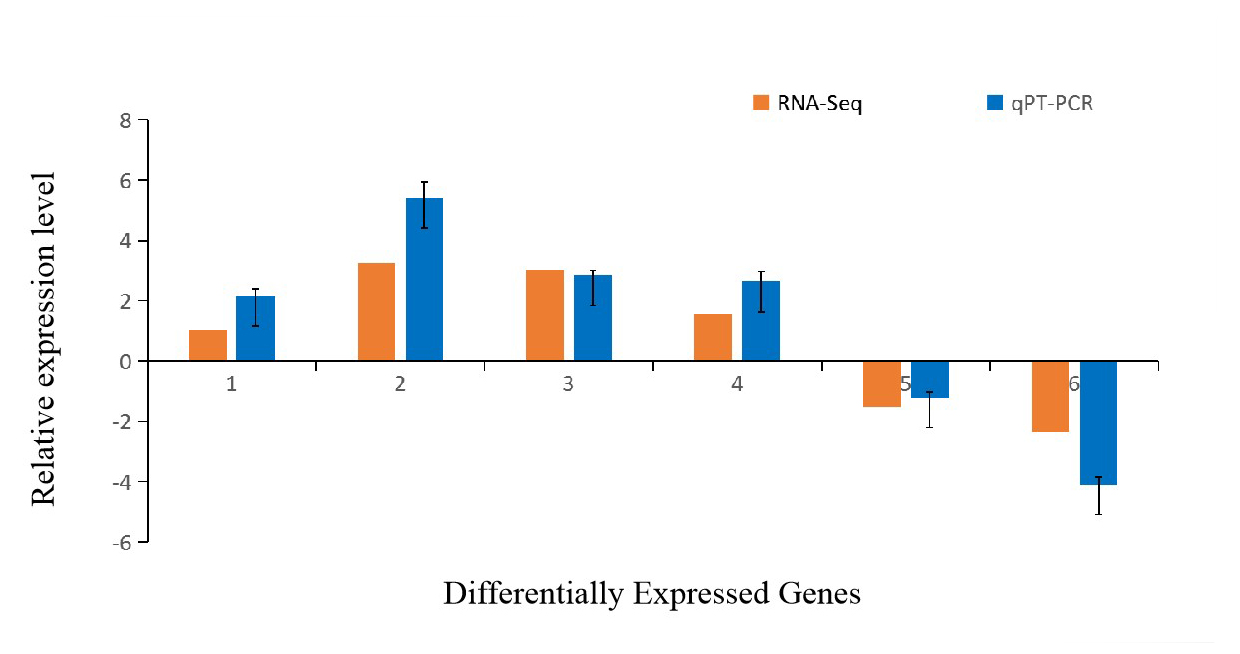
Validation of DEGs using qRT-PCR
To validate the DEGs identified by Illumina sequencing, six DEGs were chosen randomly for qRT-PCR. The results showed that the expression patterns of the selected DEGs analysis with qRT-PCR were consistent with the RNA-seq results, which indicated that the result of RNA-seq was reliable and accurate (Figure 5).

Discussion
Air exposure stress is one of the most serious stressors in cultivating and transporting crabs.27 The molecular mechanism of the response to air exposure stress is not clear. Gill is the main tissue of gas exchange and ion exchange in aquatic animals and is the first tissue to respond to air exposure stress.28 In this study, we compared the gill transcriptome of mud crab S. paramamosain with air exposure for 8h and without air exposure. 20,128 DEGs were identified, among which 13,626 were up-regulated, and 6,502 were down-regulated. DEG related to immunity, apoptosis, and ion exchange was found in this study, which is similar to those reported in previous studies.6,14,16
Air exposure stress can cause an imbalance of ion penetration.6 NKA is a key contributor to osmoregulation in crustaceans and plays an important role in the transport and regulation of osmotic ions.29 It could switch Na+ and K+ transport channels.16 NCKX is a key determinant of calcium (Ca2+) signaling and homeostasis if ion concentrations alter.30 NKCX is also the candidate’s gene to mediate this K+-dependent Na+ uptake.31 As an integral transmembrane protein, NKAT plays a key role in regulating the movement of Na+ and K+.32 NHE is a membrane protein that transports Na+ into the cells and H+ out of the cells.33 The expression of NKA, NCKX, and NKAT were upregulated, and NHE was downregulated in gills after air exposure, similar to previous research results.16 These results indicate that the gills of crabs can enhance osmotic regulation and maintain ion balance under air exposure stress.
After air exposure stress, more reactive oxygen species (ROS) will be produced in crustaceans.34 Excess ROS could damage DNA and cause apoptosis.35 To prevent or repair oxidative damage, crustaceans clear ROS with their powerful antioxidant defense system (antioxidant enzymes). Glutathione peroxidase is a kind of antioxidant enzyme that can catalyze hydroperoxide reduction.36 Thioredoxins (TRX) can clear ROS and reactivate damaged proteins through oxidative stress.37 In mammals, caspase 2 regulates apoptosis.38 When the environment is changed, caspase 2 may induce an apoptosis pathway and promote the release of cytochrome c.39 In S. paramamosain, caspase 2 induces cell apoptosis.40 In this study, the expression of glutathione peroxidase, TRX, and caspase 2 were significantly up-regulated after air exposure, indicating that ROS may accumulate in the cells and DNA may be damaged.
The immune system is usually sensitive to environmental changes. Compared with vertebrates, crustaceans do not have acquired immunity and can only rely on innate immunity to respond to environmental stress and invasion of pathogens.41 In this study, HSP70, HSP90, and HSP22 expression were up-regulated after air exposure. Hsp70 is a key factor in cellular processes and sensing environmental changes and may play a protective role in the immune system.42 HSP70 could improve the tolerance of shrimp suffering from air exposure by blocking the apoptosis pathway.35 As a molecular chaperone, Hsp90 is important in biological anti-stress.43 Hsp22 binds and degrades denatured and/or abnormal proteins, repairs misfolded proteins, maintains the homeostasis of proteins in cells, and reduces the response to harmful stimuli.44 In E. sinensis, the expression of Hsp70 and Hsp20 was also up-regulated after air exposure.6 ALFs are a host defense protein in crustaceans and against various microorganisms, including Gram-positive and negative bacteria, some fungi, and viruses.45 In E. sinensis, the expression of ALF1 was significantly increased after being stimulated by bacteria. When injected ALF1 recombinant protein into E. sinensis, the survival rate of E. sinensis was increased significantly.46 These results indicated that ALF1 is involved in the innate immunity of crabs.46 Crustins 2, an antimicrobial peptide, plays an important role in the innate immunity of crustaceans.47 Imjongjirak et al (2009) found that the crustins recombinant protein of S. paramamosain inhibited bacterial growth in vitro. As classical inhibitors of C1 cysteine protease, cystatins protected cells from adverse proteolysis of cysteine proteinases.48,49 In crabs, cystatins are potent inhibitors of papain and are involved in immune responses against invasive microorganisms.50 Cathepsin is produced by hydrolytic enzymes and sequestrated in lysosomes. Li et al.51 found that cathepsin L is involved in the innate immunity of Chinese mitten crab. The expression of cathepsin L was up-regulated after air exposure stress of E. sinensis,6 which was consistent with our research. The expression levels of these immune genes changed significantly in EG, indicating that the immune system of mud crabs was activated to respond to air exposure stress.
In this study, the top 30 KEGG pathways in which DEGs were most enriched could be roughly divided into metabolism and immunity. More materials and energy metabolism were needed for the crabs to cope with air exposure stress. The metabolism-related pathways mainly included lipids, glucose, and hormones, such as mucin-type O-glycan biosynthesis, glycerophospholipid metabolism, glucagon signaling pathway and steroid hormone biosynthesis, and so on. In the transcriptome study of hepatopancreas after air exposure of mud crab S. paramamosain, the DEGs were most enriched in the metabolism of glycolysis, lipids, and amino.14 Guo et al.1 found that L-homocysteine and lipid metabolism significantly increased in the gill of E. sinensis after air exposure. Lipids and saccharides are the main sources of energy for animals. The PI3K-Akt pathway is an important intracellular signaling pathway during the cell cycle that can reduce glycogen synthesis and increase glycolysis under hypoxia conditions.52 This study enriched DEGs significantly in ribosome pathways, which was consistent with other studies.6,16 Jiang et al.53 found that more ribosomes are generated under hypoxic conditions.53 In addition, the expression levels of cathepsins, glucosidases, sulfatase, and other enzymes in lysosome signaling pathways were significantly up-regulated after air exposure, suggesting that the lysosome pathway actively provides substances and energy to animals during air exposure. Cortisol significantly increases when fish are exposed to air.54 Most studies about crab hormones are focused on molting and gonad development, and few reports about the changes in hormone levels after air exposure.55
The changes in the environment may cause a series of innate immune responses. Previous reports on air exposure stress of crabs confirmed that immune-related KEGG was involved in air exposure.6 This study significantly enriched DEGs in three immune-related KEGG pathways: lysosome, hippo signaling, and phagosome. This result was also reported in another research about air exposure to crabs.6,16 The lysosome is an organelle degrading in eukaryotic cells and is the first barrier to innate immunity in crustaceans.56 The hippo signaling pathway consists of a series of conserved kinases that regulate innate immune response and influence the effect of immune response.57 Phagocytosis, the process by which cells internalize large particles, plays a key role in tissue remodeling, inflammation, and defense against infection caused by various microorganisms.58
Acknowledgments
Funding for this research was supported by the China Agriculture Research System of MOF and MARA (CARS-48), Guangzhou Municipal Science and Technology Project (No. 202206010138), Rural Science and Technology Correspondent Fund of Guangzhou (20212100051), the Key-Area Research and Development Program of Guangdong Province (2021B0202040001), Central Public-interest Scientific Institution Basal Research Fund, South China Sea Fisheries Research Institute, CAFS (NO.2021SD15 and 2023TD44), Sanya Special scientific research project (2020KS02) and Beihai science and technology planning project (202181013), Hainan Provincial Natural Science Foundation of China (322RC805).
Author contributions
Conceptualization: Sigang Fan (Equal), Yihui Guo (Equal). Methodology: Sigang Fan (Equal), Changhong Cheng (Equal), Xiaolin Huang (Equal), Hongling Ma (Equal). Writing – original draft: Sigang Fan (Lead), Yihui Guo (Equal). Writing – review & editing: Sigang Fan (Equal). Formal Analysis: Hongling Ma (Equal), Guangxin Liu (Equal). Supervision: Zhixun Guo (Lead). Funding acquisition: Zhixun Guo (Lead). Resources: Qibin Yang (Equal), Yougen Gao (Equal).