Introduction
Vibrio harveyi is widely distributed in the marine environment and many marine organisms, and it is an opportunistic pathogen of a variety of marine fish and invertebrates such as the pathogen of luminous vibriosis in the farmed penaeid prawns.1–3 Though the established methods of infectious disease prevention, such as immunization with vaccines, have been investigated and developed in the past ten years, control of V. harveyi in aquaculture depends majorly on antibiotics at present.4–8 But it is well known that antibiotic compounds are associated with various problems and side effects on humans and the environment.9 Drug resistance of bacteria in aquaculture became more serious.9–11
With the development of high-throughput sequencing technology, next-generation sequencing (NGS) technologies have improved the efficiency and speed of gene discovery and its function study with high throughput and low expenditure.12 RNA sequencing (RNA-Seq) can detect the expression level of thousands of genes simultaneously and deeply analyze the functional pathways and regulatory methods in biological processes.13 It can establish expression profiling and compare expression patterns between different strains or stages from the same organisms. So the technology has been widely used to discover potential drug-related genes, such as drug-resistant genes.13
Quinolone is a synthetic broad-spectrum antibacterial drug with a basic structure of the 4-quinolone ring. For example, because of their high antibacterial activity, norfloxacin and enrofloxacin are widely used to prevent and treat bacterial diseases in various aquatic animals. The resistance of bacteria to quinolone drugs in the aquaculture environment is becoming increasingly apparent.11,14 The drug-resistant mechanism of bacteria to quinolones is mainly relative to the mutation-mediated changes of the topoisomerase target site (QRDRs, Quinolone Resistance Determining Regions, specific site), plasmid (coding drug-resistance protein and efflux pump), chromosome (coding porin and efflux pump) and so on.15
At present, the resistance mechanism of quinolone has been studied on aquatic pathogens such as V. parahaemolyticus,16 Aeromonas hydrophila,17 and V. cholerae.14 QRDRs-specific site mutation and plasmid-mediated quinolone resistance genes (PMQR) qnrVC5 have been found and researched in V. parahaemolyticus.16 There were single-point mutations on GyrA and ParC in V. parahaemolyticus, which was highly resistant to ciprofloxacin. It is suggested that the mutation of the target site is an essential mechanism of drug resistance in V. parahaemolyticus.16 qnrVC5 have been found on the plasmids of four V. parahaemolyticus strains.16 In A. hydrophila, double mutations of QRDRs on gyrA and parC have also been reported, and the strains with double mutations show a high drug resistance.17 But there is a lack of research on the resistance mechanism to quinolone in V. harveyi, an important marine aquatic pathogen in China.3 In this study, transcriptome sequencing was performed on the resistant and sensitive strains of V. harveyi to obtain the changes in the transcriptome level and to disclose the relation between its drug resistance and metabolic pathway. The results can provide important data for further exploration of the molecular mechanism of quinolone resistance in V. harveyi.
Materials and Methods
Bacteria
V. harveyi wild-type strain (VS), which is sensitive to quinolone (MIC of 0.5 μg/ml), was isolated from diseased orange-spotted grouper clinically in Haikou (Hainan Province, China) and deposited at -80 °C in our laboratory, Guangdong Provincial Key Laboratory of Aquatic Animal Disease Control and Healthy Culture in China. The VS strain of V. harveyi was used to induce the drug-resistant mutant in stepwise exposure to sub-inhibitory with gradually increasing concentrations of norfloxacin (0.125, 0.25, 0.5 μg/ml, respectively) in vitro. The drug resistance and biological characteristics of strains before and after induction were detected to screen the drug-resistant mutants. Three biological replicates were performed. A high-level quinolone-resistant mutant(VR strain, MIC of 512 μg/ml)with stable resistance and cross-resistance was successfully constructed and deposited at -80 °C. Before using, bacteria strains VS and VR were cultured and activated in tryptic soy agar (TSA, pH7.5) containing 15 g/l tryptone, 5 g/l soy peptone, and 5 g/l NaCl at 28 °C for 18 h. Then, they were cultured in a TSB medium. Cells were collected for the following RNA extraction by centrifugation when OD600nm reached 0.5 (28 °C for 12 h).
RNA library construction
Total RNA was extracted following the manufacturer’s instructions using TransZol Up Plus RNA Kit (Transgen Company, Beijing) and DNA digestion with DNase I (TaKaRa, Japan). The quantity and quality of the RNA samples were measured by gel electrophoresis, Qubit 2.0, Agilent 2100, and NanoDrop 2000 (Thermo, USA). High-quality RNA of the exact quantities was used in the subsequent experiments. Three biological replicates of each group (VR and VS) were used to construct cDNA libraries.
The cDNA libraries were constructed following the instructions for the Illumina Gene Expression Sample Prep Kit (Illumina Inc., San Diego, CA, USA). In detail, the total mRNA was mixed with the fragmentation buffer and fragmented into short fragments, which were used as templates for cDNA synthesis. First-strand cDNA synthesis was carried out by random hexamer primer, and M-MuLV reverse transcriptase, and the second-strand cDNA was synthesized by DNA polymerase I and RNase H. The cDNA fragments were purified and resolved with EB buffer for end reparation and single nucleotide A (adenine) addition, and then, they were connected with adapters. The adaptor-modified fragments were selected by gel purification and amplified through PCR to create the final cDNA library.
Use Qubit 2.0 to detect the concentration of the library, and use qPCR to determine the effective concentration of the library for the library quality control. Then, high-throughput sequencing was performed on the HiSeq2500 platform (Illumina Inc., San Diego, CA, USA). Three sequencing runs used different kits, and all samples were run the same.
Bioinformatics Analysis
Clean Data, which came from raw data after removing the connector sequence and unknown and low-quality reads to avoid negatively affecting the bioinformatics analysis, were assembled to obtain the unigene library of the species. The transcripts’ structure was analyzed, and functional annotation and enrichment analysis of the differentially expressed genes (DEGs) were performed. Sequence assembly and annotation were listed in detail as follows.
De novo transcriptome assembly was performed with Trinity software.18 The resulting sequences obtained from Trinity were called unigenes.
The unigenes were compared against diverse protein databases, including the nonredundant protein (Nr) database (NCBI) and Swissprot databases (http://www.uniprot.org/blast/). The results of annotation from each protein database were presented independently. Blast2GO (http://www. blast2go.com/b2ghome) was used to obtain GO annotation of the unigenes.19 The COG database (ftp://ftp.ncbi.nih.gov/pub/COG/COG/) was also used to predict and classify functions of the unigenes.
Identification of DGEs
Gene expression levels were calculated as unit fragments per kilobase per million reads (FPKM), and the correlation between the two groups of samples was calculated. EBSeq software was used for differential expression analysis, a stringent algorithm p-value was used to identify DEGs between VR and VS strains according to the study,20 and Benjamini-Hochberg method was used to correct the significant p-value (p-value).21 The false discovery rate (FDR) was used to determine the p-value threshold in multiple tests to judge the significance of the gene expression differences. FDR≤0.05 and the absolute value of fold-change (≥2) were used as the default screening conditions to assess DEGs in this study. For GO and KEGG enrichment analyses, all differentially expressed genes were mapped to terms in GO and the KEGG database by a hypergeometric test using the whole transcriptome as the background as above.
qRT-PCR
In this study, we randomly selected 10 unigenes from the sequencing results (including resistance-related genes, recA, recN, ompU, acrB and ompA, and others, mfP, V0780, V1668, mepA and ompV) for the real-time fluorescence quantification PCR analysis (qRT-PCR) to validate the results of differential expression analysis. The sequencing results showed that fluorescent quantitative primers were designed using Primer5.0 (Table 1). cDNA was synthesized with Easy Script One-step gDNA Removal and cDNA Synthesis SuperMix in a total volume 20 μL (Total RNA (400ng/μL) 5μL, gDNA Remover 1μL, Random primer 1μL, 2×ES Reaction Mix 10μL, Easy Script RT Enzyme Mix 1μL, and RNase-free water 2μL). Firstly, the samples were incubated at 25 °C for 10 minutes, followed by a 30-minute incubation at 42°C. Finally, the samples were incubated at 85 °C for 1 minute to inactivate the enzyme. Real-time qPCR was performed with SYBR® Premix Ex Taq™ (TaKaRa, Japan) on an iQ5 Real-Time PCR system (Bio-Rad, USA) in 20 μL system (Forward Primer 0.5 μL, Reverse Primer 0.5 μL, 2 ×TransStart Tip Green qPCR SuperMix 10 μL, cDNA 2 μL and dd H2O 7 μL) under the following conditions: denaturation at 50 °C for 120 s and 95 °C for 120 s, then 40 cycles of 94 °C for 15 s, 60 °C for 15 s, and 72 °C for 60 s. Relative gene expression was analyzed by the 2−ΔΔCt method.22 gapA was chosen as the internal control for normalization. Samples were assessed in triplicate. SPSS 17.0 software was used to determine statistical significance and draw plots with Excel 2010.
Results
Sequence analysis and de novo assembly
Compared with VS, VR’s swimming ability and biofilm formation with a high-level quinolone resistance significantly increased, and the growth curve had no noticeable changes (Data not shown). The quality (concentration and RNA Integrity Number (RIN)) of RNA extracted from the above two strains is high enough to meet the requirements for the next library construction. The sequencing and assembly results are shown in Table S1 in the supplementary material, which contains 56,440,529 raw reads. Strain VS obtained 29,171,777 clean reads (91.0 % of the original data), and strain VR obtained 23,371,075 (95.9 % of the original data). Q30 base percentages are at least 88.41 %. These showed that high-quality clean data was obtained, ensuring the accuracy of sequence assembly and subsequent analysis. FPKM value (closer to 1) also indicated a better reproducibility of the two groups.
The total transcriptome data (NCBI accession number PRJNA791968) was spliced de novo, and then assembled and get a total of 2,082 unigenes with a total length of 4,687,723 bp and an average length of 2,251 bp. The number with a length between 200 and 300 bp is the largest, in total 745 and accounting for 35.78 %. And the unigenes with more than 3,000 bp is 379, accounting for 18.2 % (Figure S1 in the supplementary material).
Sequence annotation
Using Glimmer soft to perform gene prediction on the unigene library, a total of 2, 519 genes and their CDS sequences were obtained, and the average length of the predicted genes was 1, 190 bp. The predicted genes were annotated functionally, and finally obtain 2, 509 genes with the annotated information (Table S2 in the supplementary material).
Differentially expressed genes
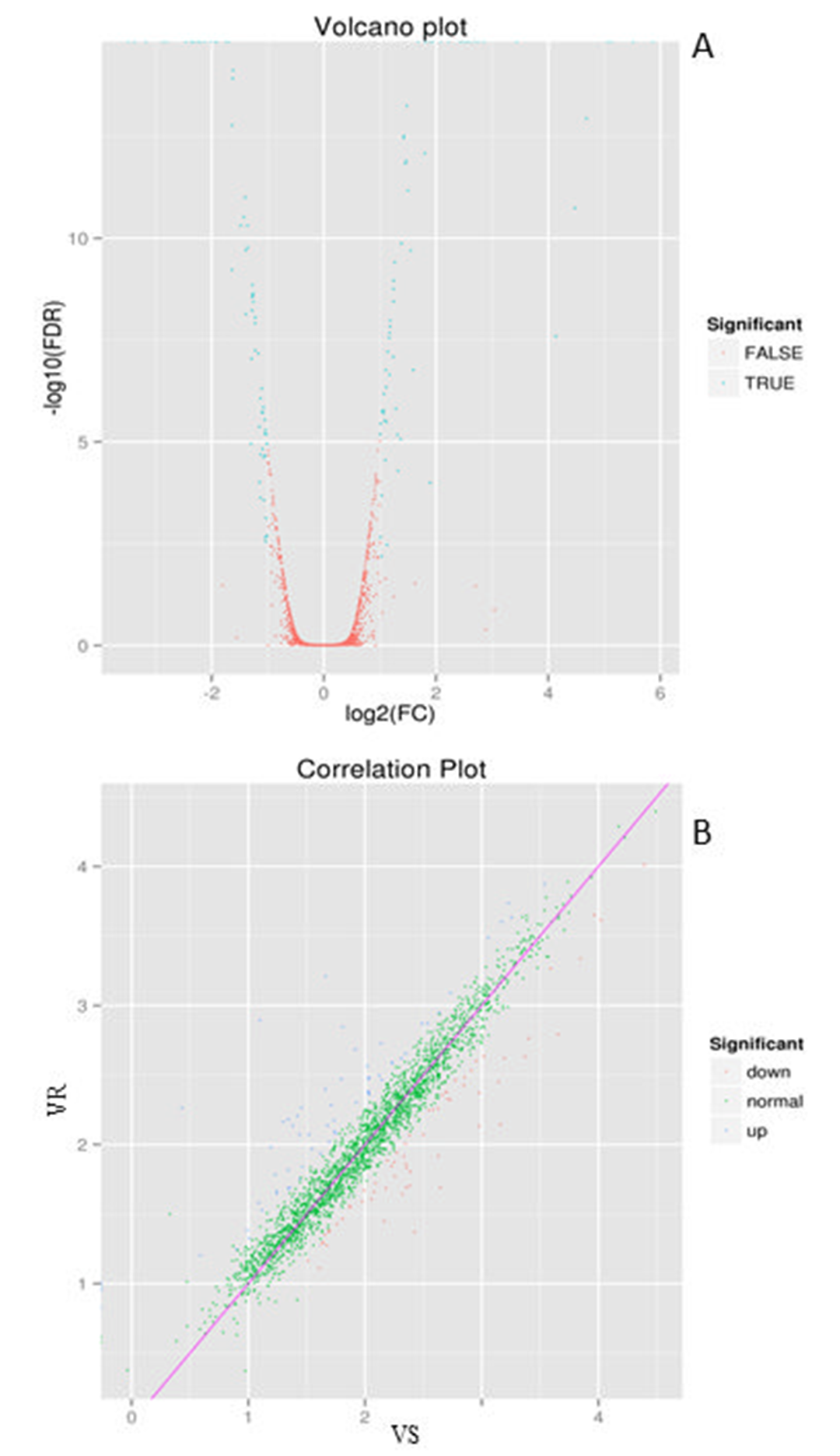
129 DEGs were found finally. Compared with strain VS, drug-resistant mutant VR had 65 genes up-regulated and 64 genes down-regulated, of which 5 genes were only expressed in drug-resistant bacteria. The volcano diagram of DEGs was shown in Figure 1A. DEG correlation scatter plot was shown in Figure 1B.

Enrichment analysis of DEGs
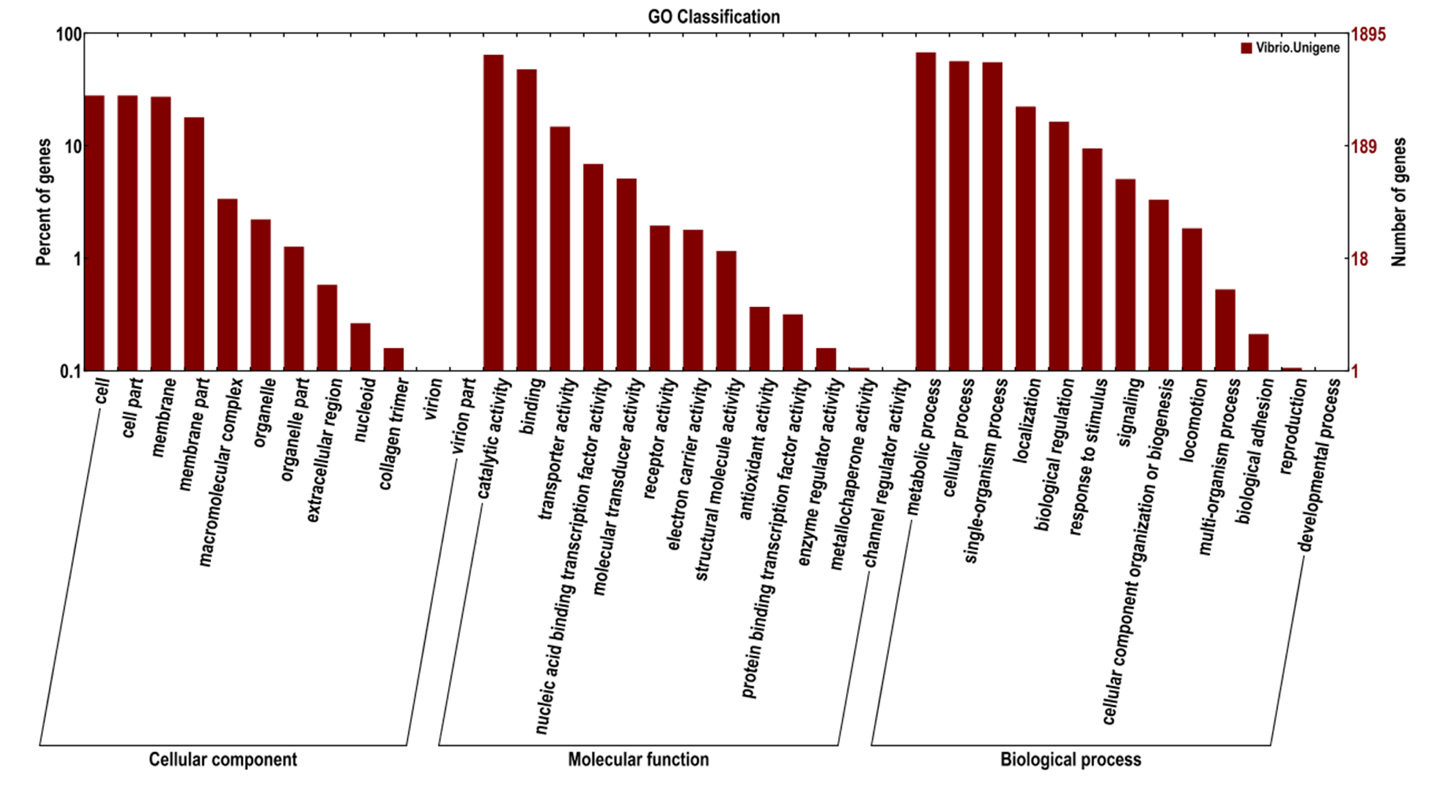
DEGs between strain VS and VR and the annotated results of all genes at the GO secondary node are shown in Figure 2. The results showed that all unigenes are annotated into 38 second-level entries, of which DEGs are annotated into 26. Regarding molecular functions, they mainly focus on transporter activity and binding. For cell composition, major categories with differences were cell (includes the plasma membrane and any external encapsulating structures such as the cell wall and cell envelope), membrane, and cell part (any constituent part of a cell). Among biological processes, metabolic processes and cellular processes were the most represented. The results show that drug-resistant bacteria differ from sensitive bacteria in cell structure, substance metabolism, and transport.
.png)
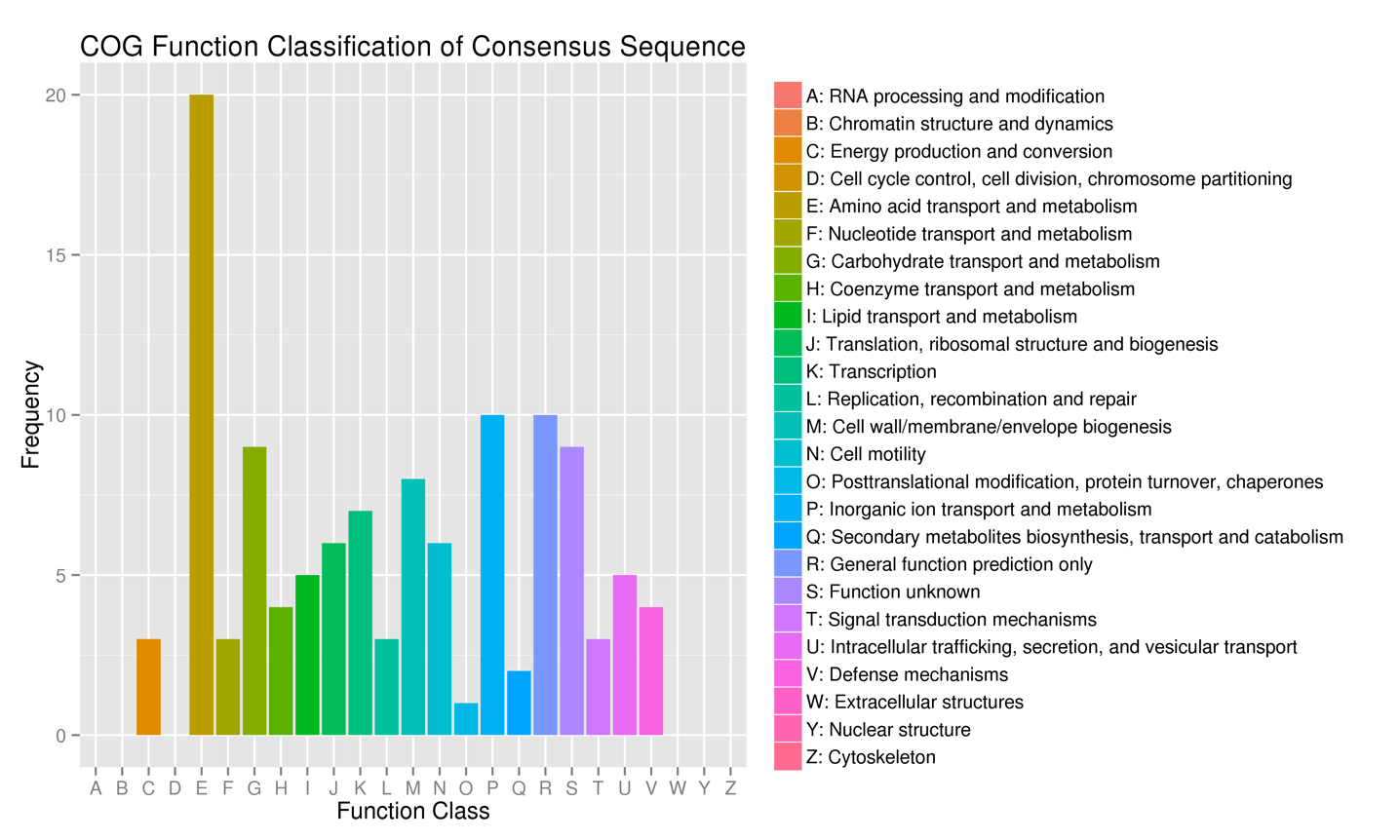
The function of these DEGs was predicted by comparing them with COG databases. The statistical results of COG classification of the DEGs are shown in Figure 3. 90 DEGs were mapped to 19 different functional categories, which mainly focused on amino acid transport and metabolism, cell wall/membrane biosynthesis, carbohydrate transport and metabolism, ribosomal structure, and biosynthesis.
.png)
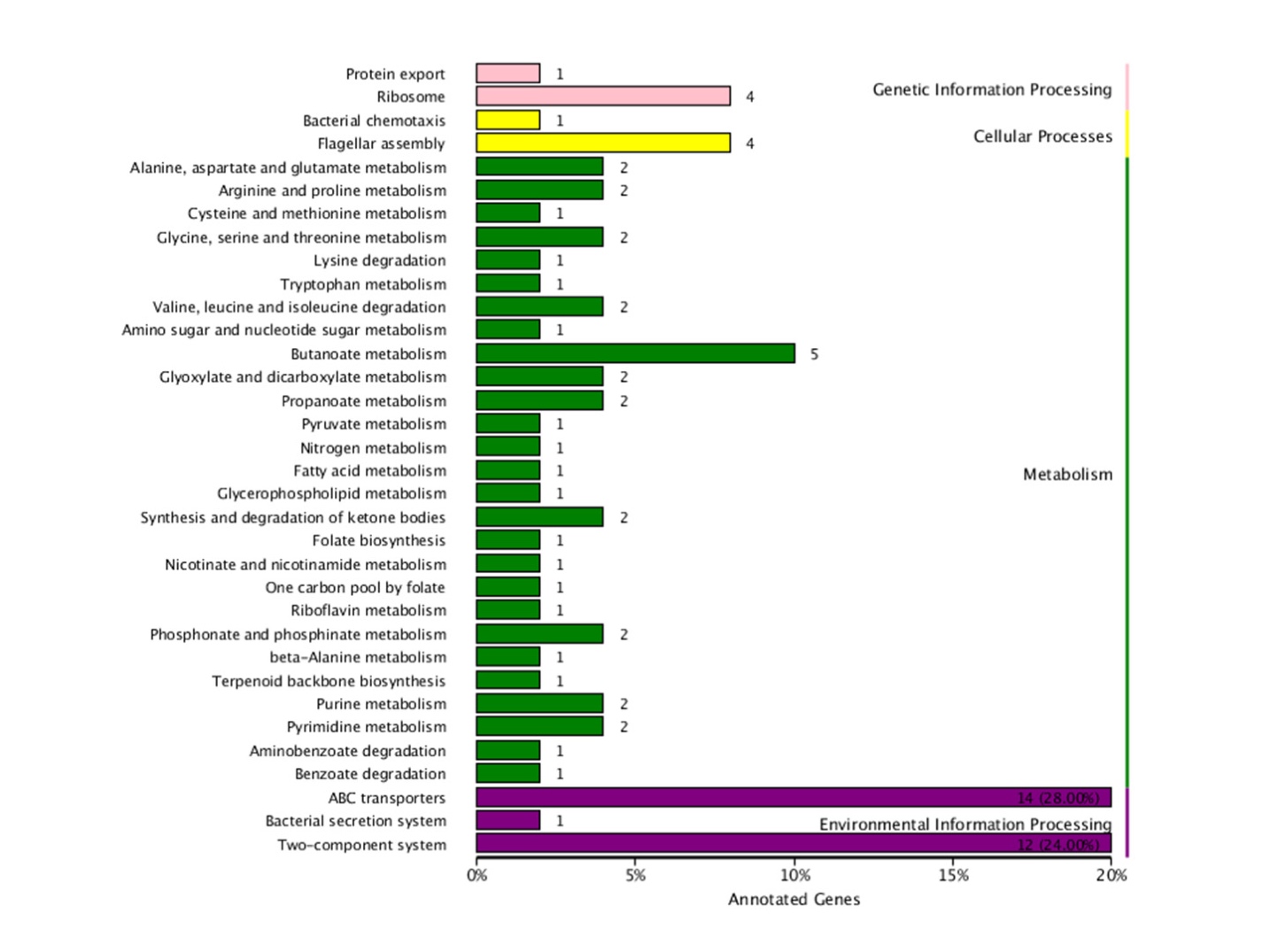
The enrichment degree of KEGG pathway was analyzed using the enrichment factor, and Fisher’s test was used to calculate the significance of enrichment. 78 DEGs were annotated to 34 metabolic pathways. KEGG classification map is shown in Figure 4, and Table 2 shows the enriched top 20 channels. The results showed that the pathway two-component system (ko02020), ABC transport system (ko02010), and flagella assembly (ko02040) have the largest number of the annotated differential genes, and they are enriched significantly (Figure S2-S4 in the supplementary material).

Analysis of the resistance-related genes
Some genes related to quinolone resistance were identified from the DEGs, including resistance-related transport proteins, outer membrane proteins, and DNA repair-related proteins (Table 3). The results showed that 5 drug resistance transporter genes were up-regulated, among which resistance protein B (BSP001668) was up-regulated most. Genes of two outer membrane proteins (OmpA and OmpU) are down-regulated. The expression levels of DNA repair protein RecN and DNA recombinant protein RecA are both up-regulated. Since the target of quinolones is topoisomerase in cells, proteins related to DNA repair may play an indirect role in its drug resistance.
Results with fluorescence quantification PCR
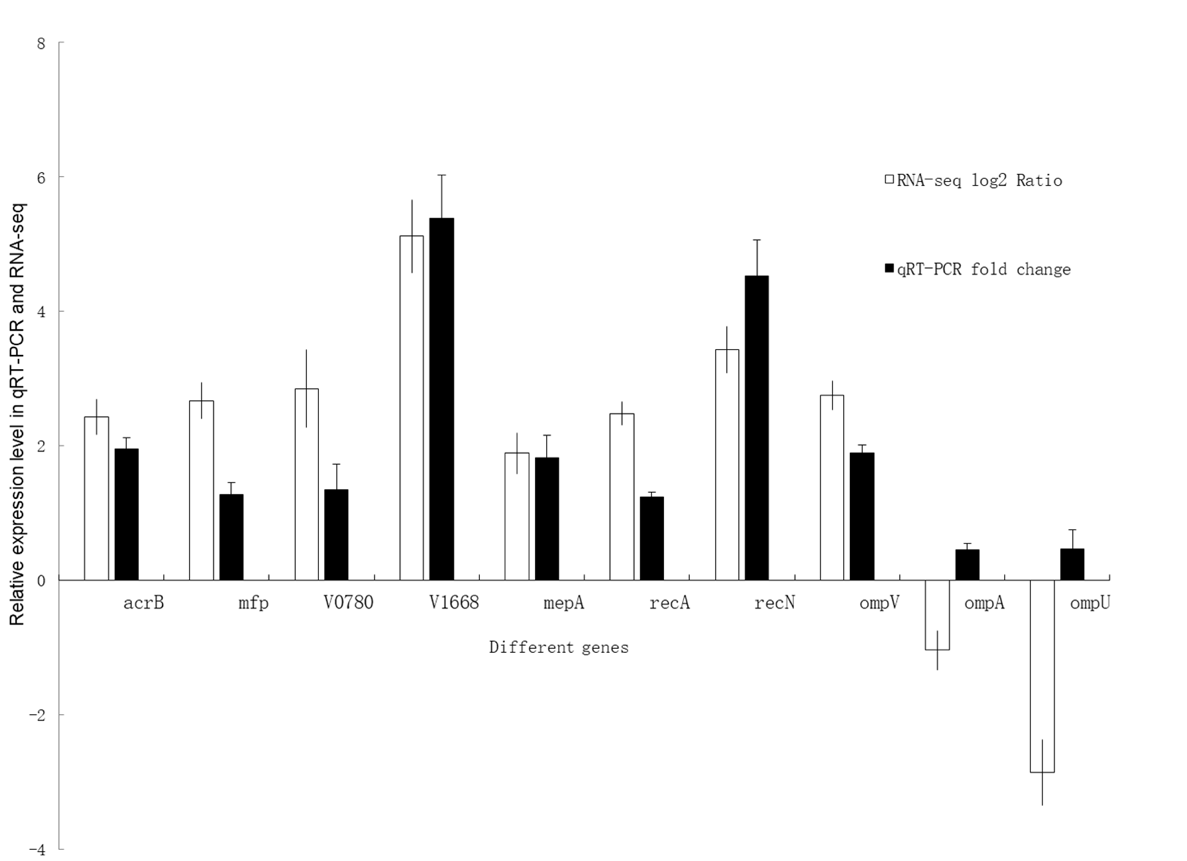
Ten genes were selected from the transcriptome data, and their fluorescence quantitative analysis was performed (Figure 5). The expression level of 8 genes, except genes ompA and ompU increased differently. Of which, gene V1668 and recN increased with the most degree. The fold changes of fmp, V0780, and recA are also more than 1. The results showed that the expression changes of these genes are consistent with the transcriptome analysis results. Overall, the transcriptome sequencing data in this study is reliable.

Discussion
The resistance mechanism of Vibrio to antibiotics is very complex.15 The single-drug induction method adopted in this study can exclude the interference of the other environmental factors in the bacteria resistance. So the obtained drug-resistant strain VR can be used as a single sample induced by quinolone.
The next-generation high-throughput sequencing technology RNA-Seq was used for transcriptome sequencing analysis of quinolone-sensitive and induced drug-resistant strains of V. harveyi in the study. A total of 13.23 Gb Clean Data was obtained, providing sufficient and adequate data and information for the following research (Table S1 in the supplementary material). The Assembly de novo technique was used to obtain the transcripts of the species, and a unigene library of the species was constructed, which obtained a total of 2,082 unigenes (Figure S1 in the supplementary material). In this study, there were 129 differentially expressed genes, 65 of which were up-regulated and 64 were down-regulated in mutant strain VR (Figure 1). But Li et al.24 analyzed the transcriptome of ciprofloxacin-induced Salmonella typhimurium ATCC13311-resistant strains. They obtained 3,466 differentially expressed genes compared with the sensitive bacteria, of which 3,434 were up-regulated. The down-regulated genes had only 32 genes. This result may be due to the differences in species and to the different drugs. It also showed that it is necessary to study further the action mechanism of drugs and the drug resistance mechanism of the different bacteria.
The annotations of unigenes can provide gene functions, physiological processes, and pathways involved in Vibrio.13 Combining the functional annotation and enrichment analysis results of differentially expressed genes and the resistance-related genes analysis result, DEGs of the two-component system (TCS), flagella assembly, active efflux system, outer membrane proteins, DNA repair system, and cell membrane structure were discovered in quinolone-resistant V. harveyi (Figure 2-5). But QRDRs and qnrVC5 have not been found, like in V. Parahaemolyticus.16
There are 12 differentially expressed genes annotated to the two-component system pathway (ko02020) (Figure 4 and Table 2). TCS is a signal transduction system for bacteria responding to environmental changes.25 It regulated multiple functions of bacteria, including bacterial growth, biofilm-forming, drug resistance, virulence, quorum sensing, etc.26 Among the up-regulated genes, it was found that the fliC (BSP000991) gene, relating to the assembly of flagellar (Table 2), was regulated by a two-component system. Flagella assembly plays a significant role in bacterial drug resistance.25 Transcriptome results showed that the expression of flagella-related proteins increased in the drug-resistant Vibrio. This is consistent with our previous research results that the phenotypic characteristics of drug-resistant bacteria, such as their mobility and biofilm formation ability, were increased (data not shown). QseBC, some TCS of E. coli, also participated in the transcriptional regulation of flagellar regulation-related genes (flhD, flhC, fliA, motA, fliC).26 Flagellar expression increased in the present study, and as a result, the drug-resistant strain exhibited increased mobility and biofilm formation. So the regulation function of TCS on the flagella-related proteins is very important to drug-resistance of bacterium. This may be an indirect and non-specific drug-resistance mechanism for V. harveyi like that in E. coli.
Active efflux system is important in bacterial multi-drug resistance.27 In our results, four drug resistance-related differentially expressed transporters, AcrB (BSP000778), RND family efflux transporter (BSP000779), resistance protein (BSP000780), and resistance protein B (BSP001668) belongs to the RND efflux pump family of active efflux system. Multidrug transporter MatE (BSP002444) belongs to the MATE family (Table 2 and Table 3). The RND-type efflux pump AcrAB-TolC system is an active efflux system in E. coli. Its related substrates include quinolones, β-lactams, macrolides, chloramphenicol, tetracycline and other antibiotics.28 MATE transporter YdhE of E. coli can also transport quinolones and macrolide drugs.29 V. parahaemolyticus NorM, mediating resistance to quinolone drugs, has 57 % identity and high similarity (88 %) with E. coli YdhE and belongs to the MATE family of transporters.27 Our results showed that the resistance of V. harveyi to quinolone drugs is likely related to the efflux pump system.
The outer membrane’s permeability also affects bacteria’s sensitivity to quinolones.30 It has been found that the outer membrane proteins of E. coli OmpF and OmpC are related to the diffusion of quinolone drugs. Hydrophilic drugs (ciprofloxacin, norfloxacin) diffuse mainly into bacteria through OmpF.30 The expression of two outer membrane proteins (OmpA and OmpU) decreased in our study (the relative expression level < 0) (Table 3), of which OmpA and E. coli OmpF have a high homology. Decreased expression of outer membrane proteins can reduce the diffusion of quinolone drugs and enable bacteria to obtain drug resistance. So outer membrane proteins (OmpA and OmpU) can be related to the quinolone resistance of V. harveyi.
Because real-time fluorescent quantitative PCR has high sensitivity and reproducibility, it can detect small differences in gene expression, and it can be used to measure the expression level of genes and verify the results of transcriptome analysis and their functions in drug resistance of V. harveyi.22 In this study, the specific primers and the reaction system with good melting curve were selected. The housekeeping gene gapA in bacteria was selected as the internal reference gene for qPCR after the reaction conditions and system, and the primers were optimized. qPCR results are consistent with the transcriptome analysis such that the expression of ompA and ompU also decreased (the relative expression level < 1), and the expression of the other genes also increased (Figure 5), confirming the reliability of transcriptome sequencing and data analysis, and confirming that the above-related genes, acrB, ompA, ompU, recA and recN, may play key roles in the drug resistance of V. harveyi.
The metabolic pathways of two-component systems, flagella assembly, active efflux system, and outer membrane proteins, are significantly different, suggesting that changes in various bacterial metabolism pathways can lead to drug resistance. This is an important supplement to the study on drug resistance mechanisms though QRDRs and differentially expressed qnrVC5 have not been found in V. harveyi. Results provide essential data for exploring the molecular mechanism of quinolone resistance in V. harveyi and present a great reference to unravel the mechanism of quinolone resistance in the other different Vibrios. The relationship between the drug resistance and the metabolic pathway deserves further exploration.
Conclusion
This study compared V. harveyi transcriptome profiles between the wild-type strain and its quinolone-resistant mutants. Comparative transcriptome analyses identified 9 genes associated with quinolone-resistance ability, some further verified by qPCR. The resistance mechanism of V. harveyi to quinolones is related to multiple pathways. The two-component system, flagella assembly, active efflux system, outer membrane protein, and DNA repair system constitute a complex network in developing drug resistance of V. harveyi, and their relation or co-operation between any two need to be studied further. Taken together, our results provide enough information for quinolone-resistance study of V. harveyi. And also provides a reference for the further research on the physiological changes and metabolic processes in the resistance formation of Vibrios.
Acknowledgments
Thank all the people who have dedicated time to the experiments. This work was supported by grants from the Natural Science Foundation of Guangdong Province (2022A1515012203) and the National Key Technology Support Program of China (No. 2012BAD17B03).
Authors’ Contributions per CRediT
Conceptualization: Xiaochen Tang (Equal), Yu Ding (Equal). Methodology: Xiaochen Tang (Equal), Yu Ding (Equal). Formal Analysis: Xiaochen Tang (Lead). Writing – original draft: Xiaochen Tang (Lead). Writing – review & editing: Xiaochen Tang (Equal), Yu Ding (Equal). Funding acquisition: Yu Ding (Lead). Resources: Yu Ding (Lead). Supervision: Yu Ding (Lead).